1D semantic segmentation
T.O.C.
- 1D semantic segmentation
- Code
- Training and data
- Results
- The good
- The bad
- The confusing
- Discussion
Context
Semantic segmentation is an extension of image segmentation, whereby the partitioned pixels are associated with labels to provide meaning to each subset of pixels.
It is apparent that as semantic segmentation was devised for images most of its approaches are 2D-centric e.g. 2D convolutional neural networks (CNN), converting the pixels into a $k$-nearest-neighbors (KNN)-graph and applying graph theory, etc.
While the focus on 2D is not prohibitive of 1D applications, improvements in the field which stem from leveraging the structure of images may not translate well into any given 1D use case.
So when might the need for 1D semantic segmentation arise, and how can it be applied?
"1D"
Foremost let us clarify "1D". In this context we refer to "1D" similarly as to how images are "2D" i.e. we view the image with a given width ( $w$, $d_1$) and height ( $h$, $d_2$), but images also consist of at least 1 (and commonly 3) channels ( $c$, $d_3$) conveying color information (thus $w\times h \times c$).
Thus "1D" is a multi-channel sequence and semantic segmentation is analogous in aim to compressive sensing or detection theory.
Example
An example of when 1D semantic segmentation may be preferable to traditional signal detection methods can be found in biology. Two common sequences found in biology are those of amino-acids and those of DNA of which I will be considering the latter for this post. DNA - from a sequence standpoint - is (generally) made up of four characters (nucleotides or NT): A, C, T, and G. Further, a DNA sequence can be encoded - via one-hot encoding - into an $n\times4$ matrix.
While the $n\times4$ matrix could be treated as a single channel image ( $n\times4]times1$), for $n>>4$ the common set-up of $3\times3$ convolutional kernels (for 2D-CNNs) makes less sense.
As for using a Recurrent Neural Network (RNN) such as something with a Long-Short Term Memory (LSTM) module, given that the human genome is over 3 billion NT, that doesn't work so well either if we leave the sequence unaltered.
Thus it makes more sense to break up the sequence into fixed-length chunks (e.g. 300 NT), where the length chosen is large enough to contain the features one wishes to label. These chunks could be fed into a LSTM, but here we will continue exploring 1D semantic segmentation.
Code
For 1D semantic segmentation, I started by adapting the Ademxapp Model A1 Trained on Cityscapes Data architecture:
ReferenceModel = NetModel["Ademxapp Model A1 Trained on Cityscapes Data"]
the main components of which seem to be residual convolutional blocks of one of two main forms:
form 1: 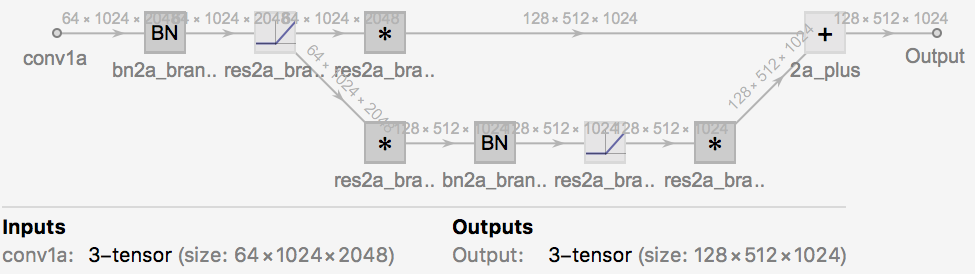
form 2: 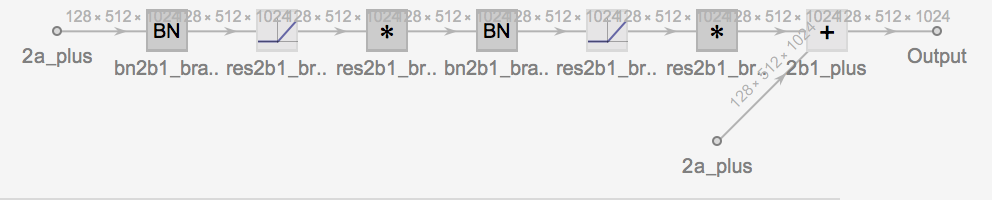
Which we can recreate with:
AdemxappResidualCNN1DBlock[inputDimension_, name_, residualBNQ_: True, kernel_: 3, padding_: 1]:=
With[
{ scale = If[residualBNQ, 2, 1] },
Module[
{
residualBranch = { NetPort["Input"] -> name <> "_plus_shared" },
residualBNBranch = {
name <> "_ramp_shared" -> name <> "_cnn1d_branch_1" -> name <> "_plus_shared"
},
mainBranch = {
name <> "_bnl_shared" -> name <> "_ramp_shared",
If[residualBNQ,
name <> "_ramp_shared" -> name <> "_cnn1d_branch_main" -> name <> "_ramp_branch_main",
name <> "_ramp_shared" -> name <> "_cnn1d_branch_main" -> name <> "_bnl_main" -> name <> "_ramp_branch_main"
],
name <> "_ramp_branch_main" ->
name <> "_cnn1d_branch_main_a" -> name <> "_plus_shared"
},
residualBranchLayers = <||>,
residualBNBranchLayers = <|
name <> "_cnn1d_branch_1" -> ConvolutionLayer[scale*First@inputDimension, {1}]
|>,
mainBranchLayers = <|
name <> "_bnl_shared" -> BatchNormalizationLayer["Input" -> inputDimension],
name <> "_ramp_shared" -> Ramp,
name <> "_cnn1d_branch_main" -> ConvolutionLayer[scale*First@inputDimension, {kernel}, "PaddingSize" -> {padding}],
If[residualBNQ, Nothing, name <> "_bnl_main" -> BatchNormalizationLayer[]],
name <> "_ramp_branch_main" -> ElementwiseLayer[Ramp],
name <> "_cnn1d_branch_main_a" -> ConvolutionLayer[scale*First@inputDimension, {kernel}, "PaddingSize" -> {padding}],
name <> "_plus_shared" -> ThreadingLayer[Plus]
|>,
layers, connections
},
If[residualBNQ,
layers = Join[mainBranchLayers, residualBNBranchLayers];
connections = Join[mainBranch, residualBNBranch],
layers = Join[mainBranchLayers, residualBranchLayers];
connections = Join[mainBranch, residualBranch]
];
NetGraph[layers, connections]
]
]
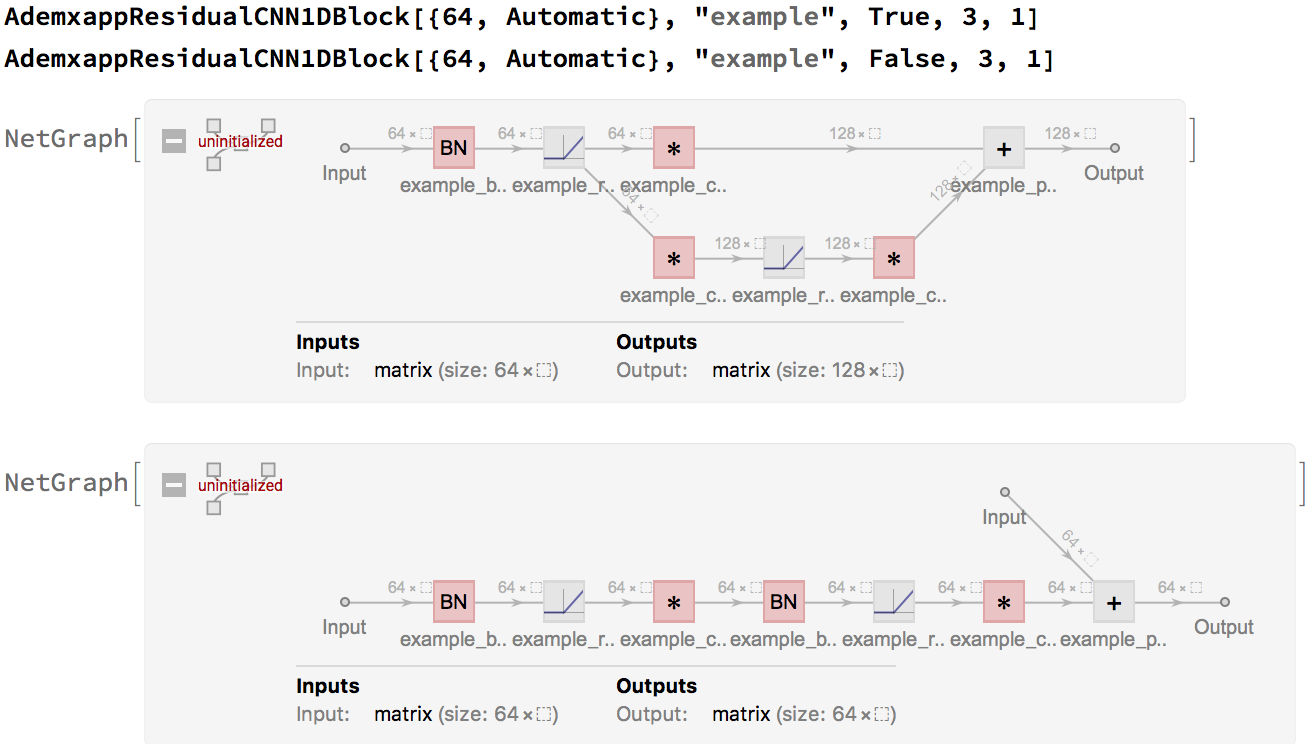
Since these are often used in a chain, in the form of form1->form2->form2->form2, a simple helper, will tidy up our model:
AdemxappResidualCNN1DBlockChain[inputDimension_, name_, length_: 1, kernel_: 3, padding_: 1]:=
NetChain[
Join[
{
AdemxappResidualCNN1DBlock[inputDimension, name, True, kernel, padding]
},
With[
{
newInputDimension = {2 First@inputDimension, Last@inputDimension}
},
Table[
AdemxappResidualCNN1DBlock[newInputDimension, name, False, kernel, padding]
, {length - 1}
]
]
]
]
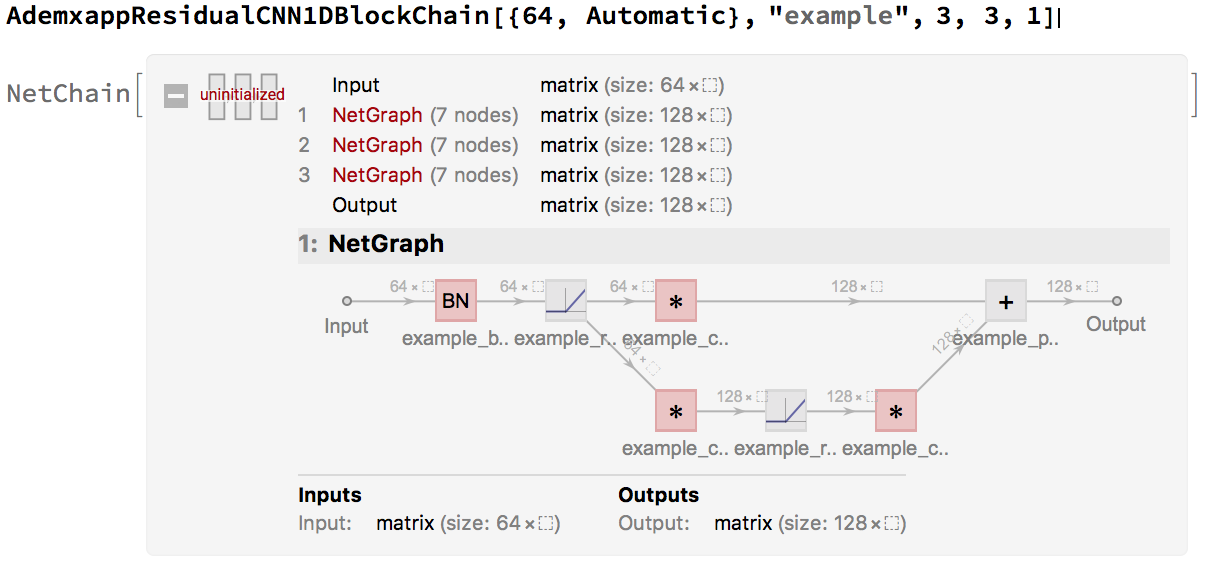
While the chain is nice, since $n>>4$ we will use an inception model. With the following helper:
PadToPreserveLength[kernel_] := Floor[kernel/2]
We can created a sequence length preserving inception architecture as follows:
AdemxappResidualCNN1DInceptionBlockChain[inputDimension_, name_, length_: 1, kernels_: {}] :=
NetGraph[
Join[
(* Residual chain for each kernel *)
Table[
name <> "_" <> ToString[k] ->
AdemxappResidualCNN1DBlockChain[inputDimension, name, length, k, PadToPreserveLength[k]]
, {k, kernels }
],
(* Pooling layer for each kernel *)
Table[
name <> "_pool_" <> ToString[k] ->
PoolingLayer[{k}, "PaddingSize" -> PadToPreserveLength[k]]
, {k, kernels }
],
(* Batch norm for input and thread to combine output *)
{
name <> "_batch_norm" -> BatchNormalizationLayer[],
name <> "_thread" -> ThreadingLayer[Plus]
}
],
Table[
name <> "_batch_norm" -> name <> "_" <> ToString[k] ->
name <> "_pool_" <> ToString[k] -> name <> "_thread"
, {k, kernels}
]
]
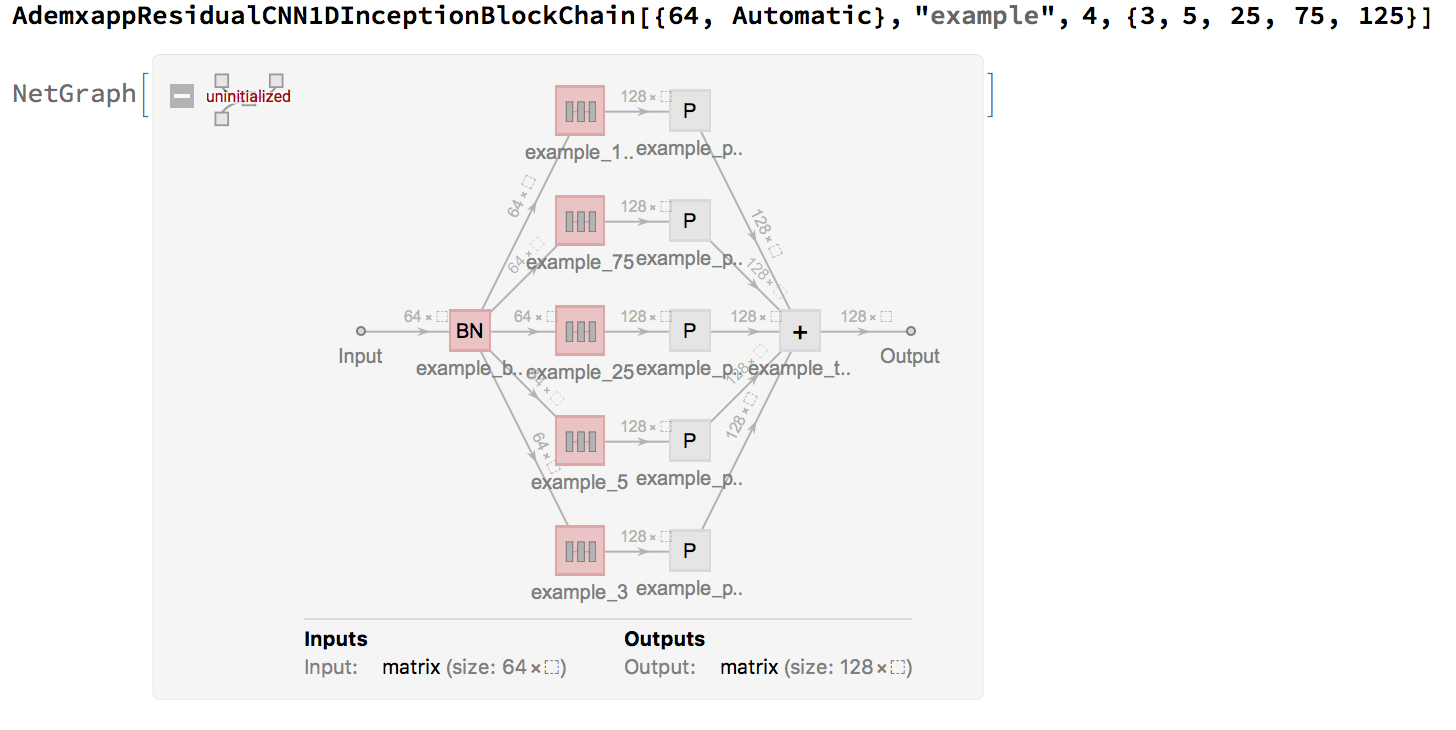
This makes writing our large model much cleaner and easy to modify:
NetGraph[
<|
"input_cnn" -> ConvolutionLayer[64, {3}, "PaddingSize" -> {1}],
"input_pool" -> PoolingLayer[{3}, "PaddingSize" -> {1}],
"incept_1" ->
AdemxappResidualCNN1DInceptionBlockChain[{64, Automatic},
"incept_a", 3, {3, 25, 75}],
"ramp_1" -> Ramp,
"incept_2" ->
AdemxappResidualCNN1DInceptionBlockChain[{128, Automatic},
"incept_b", 3, {3, 25, 75}],
"ramp_2" -> Ramp,
"incept_3" ->
AdemxappResidualCNN1DInceptionBlockChain[{256, Automatic},
"incept_c", 3, {3, 25, 75}],
"ramp_3" -> Ramp,
"incept_4" ->
AdemxappResidualCNN1DInceptionBlockChain[{512, Automatic},
"incept_d", 3, {3, 25, 75}],
"ramp_4" -> Ramp,
"dsc2" -> ConvolutionLayer[256, {3}, "PaddingSize" -> {1}],
"drmp2" -> Ramp,
"dsc4" -> ConvolutionLayer[64, {3}, "PaddingSize" -> {1}],
"drmp4" -> Ramp,
"pcnn" -> ConvolutionLayer[3, {3}, "PaddingSize" -> {1}],
"trsp" -> TransposeLayer[],
"sfmx" -> SoftmaxLayer[]
|>,
{
"input_cnn" ->
"input_pool" ->
"incept_1" ->
"ramp_1" ->
"incept_2" ->
"ramp_2" -> "incept_3" -> "ramp_3" -> "incept_4" -> "ramp_4",
"ramp_4" ->
"dsc2" -> "drmp2" -> "dsc4" -> "drmp4" -> "pcnn" -> "trsp" -> "sfmx"
}]
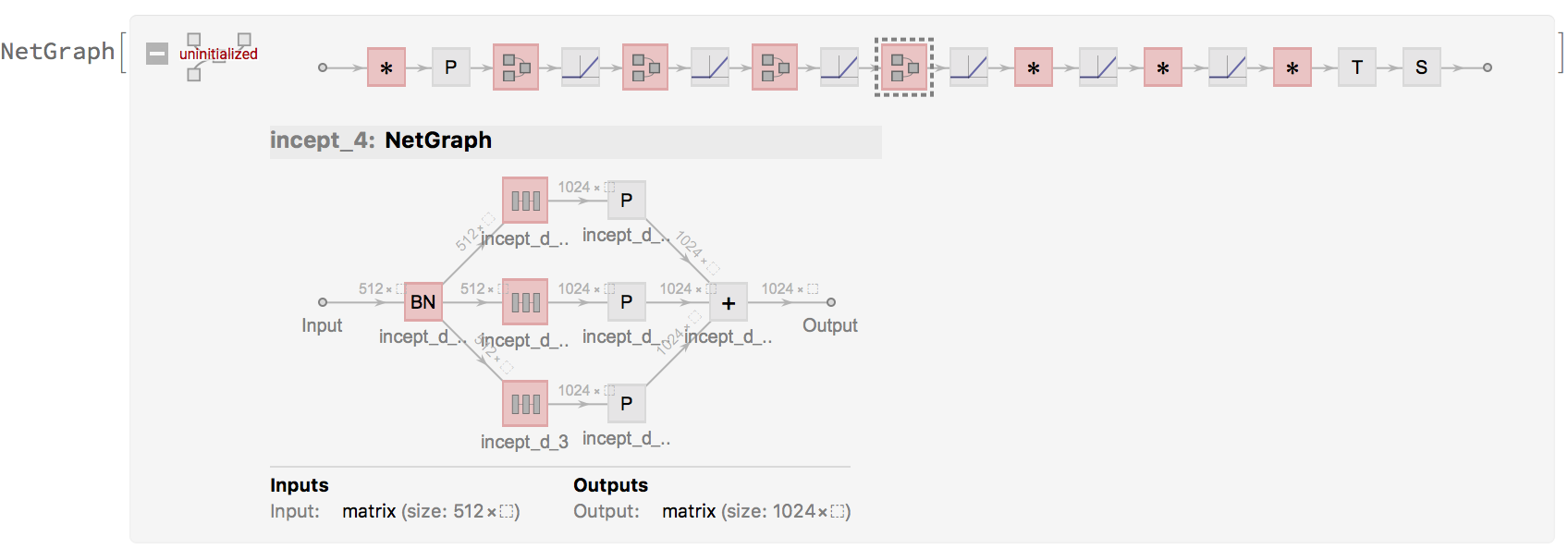
which could now actually be simplified with NetChain :)
Training and data
The data used here is from the annotated human genome hg38.fa. We will be searching for exonal, intronal, and "other" sequences. Since the main prerogative is finding exonal sequences, and most exons are in the range $\left{50, 250\right}$, we will use DNA sequences of $300$NT.
Note, the output of the model above is $300\times3$ matrix. This is the class probabilities for each NT (exon, intron, other), since we are using soft labeling. Training this model took $48$ hours on AWS.
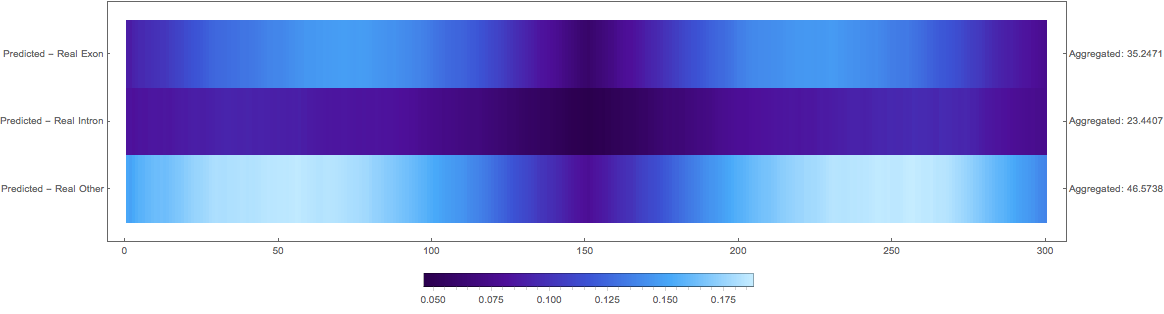
Results
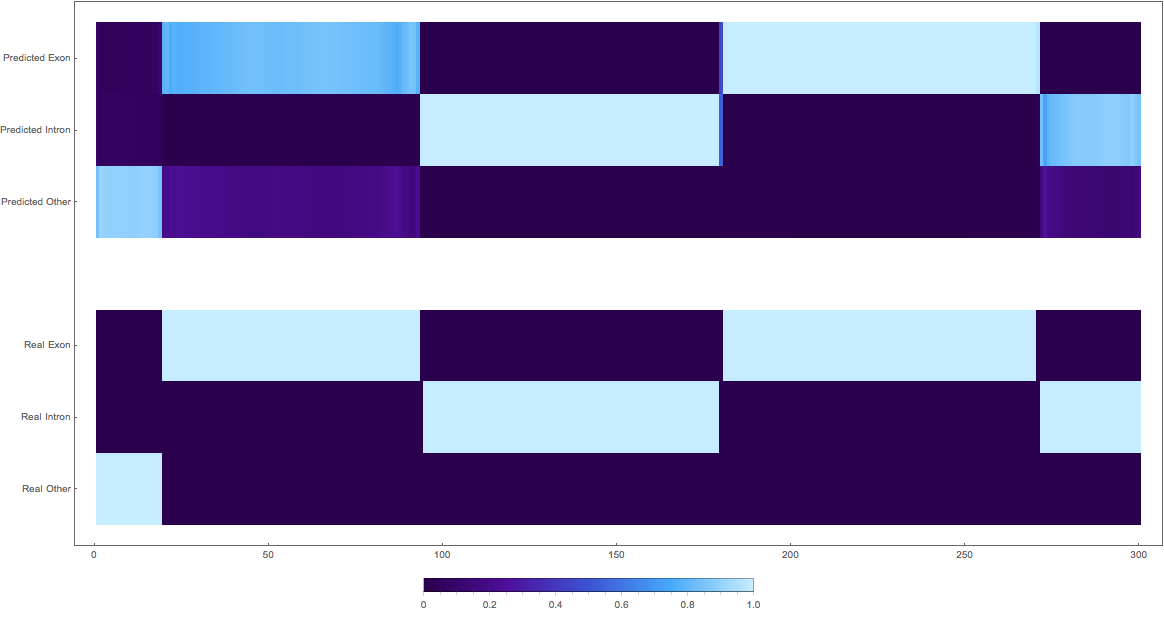
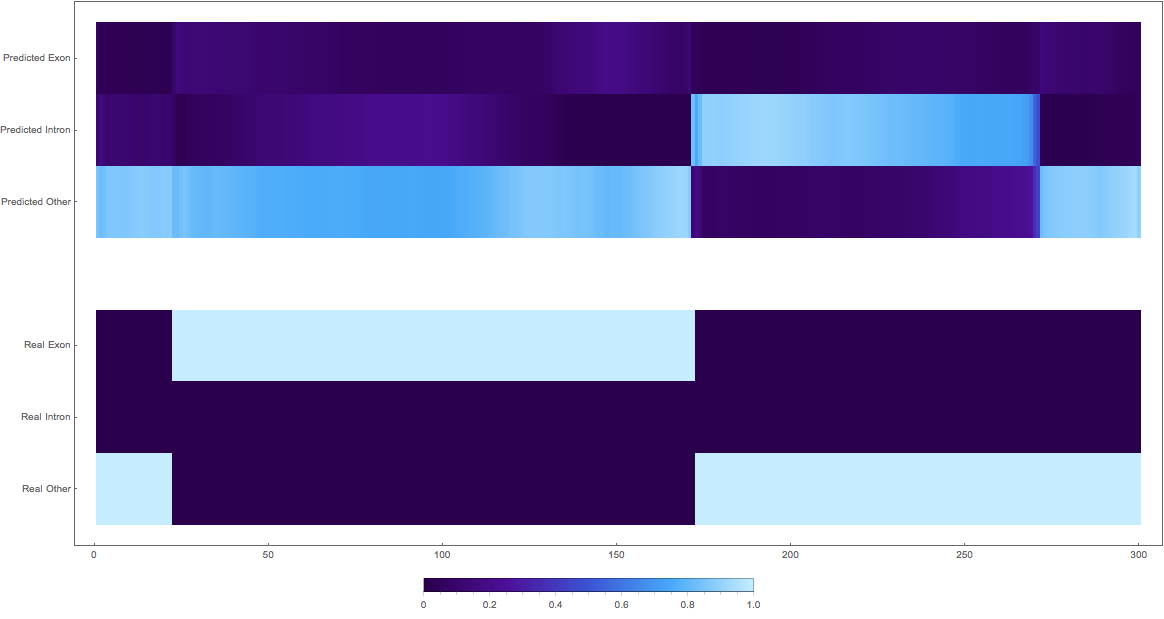
In the images below the top three rows of the ArrayPlot belong the probabilities predicted by the network, where the bottom three are the "ground truth"
The Good
1:
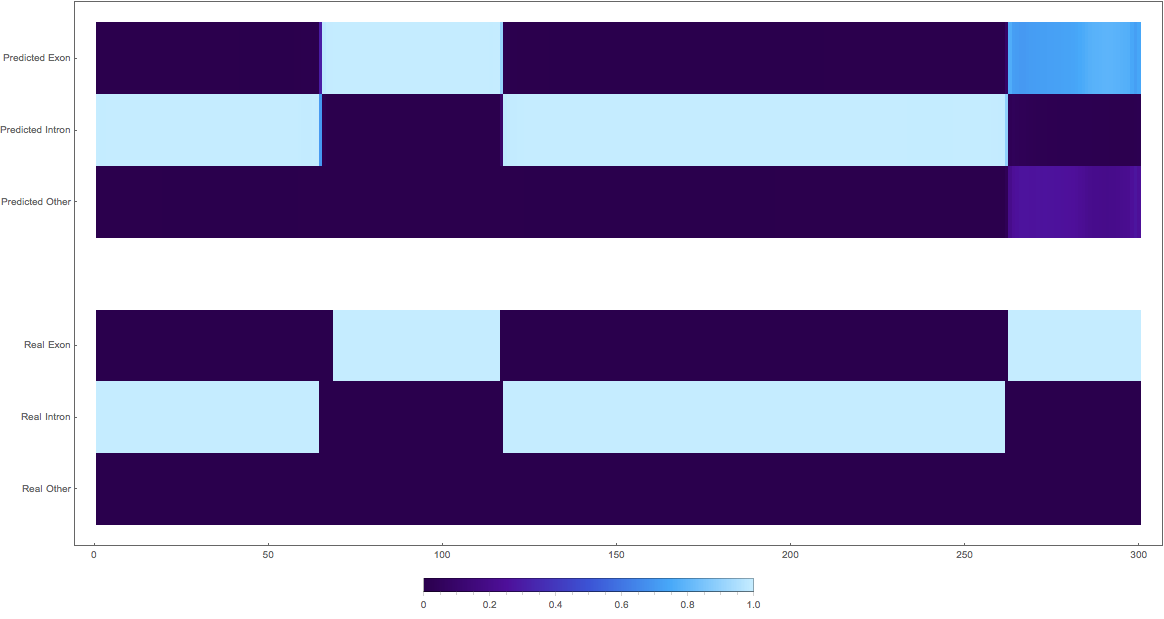
2:
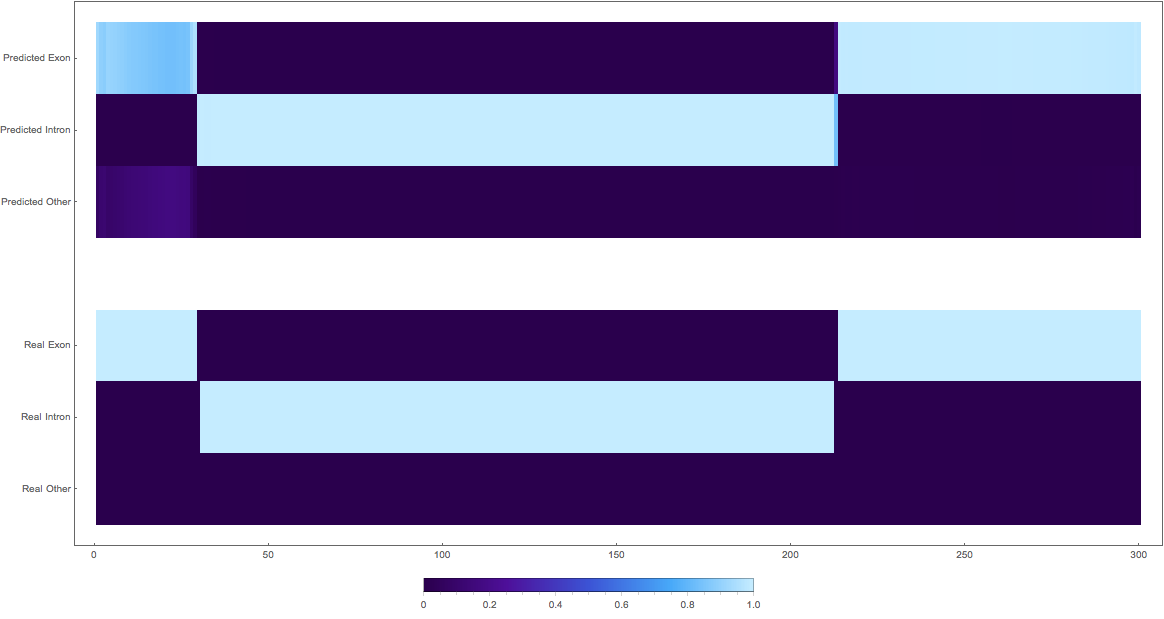
3: (not so great, but interesting)
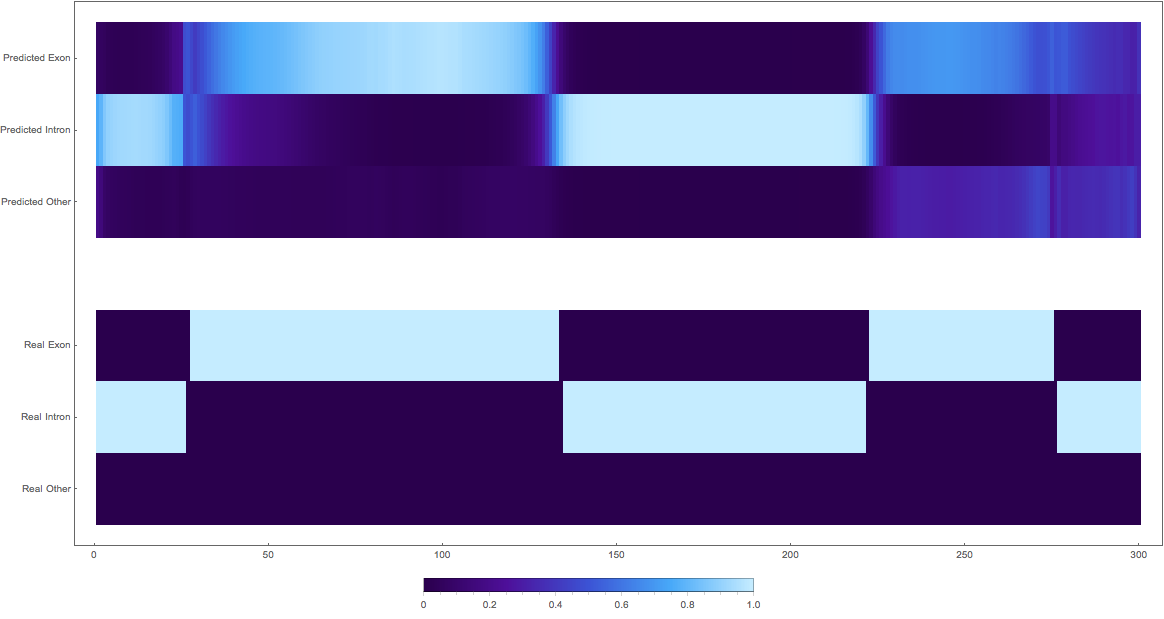
The Bad
1. (might actually be good as some exons are not yet annotated) 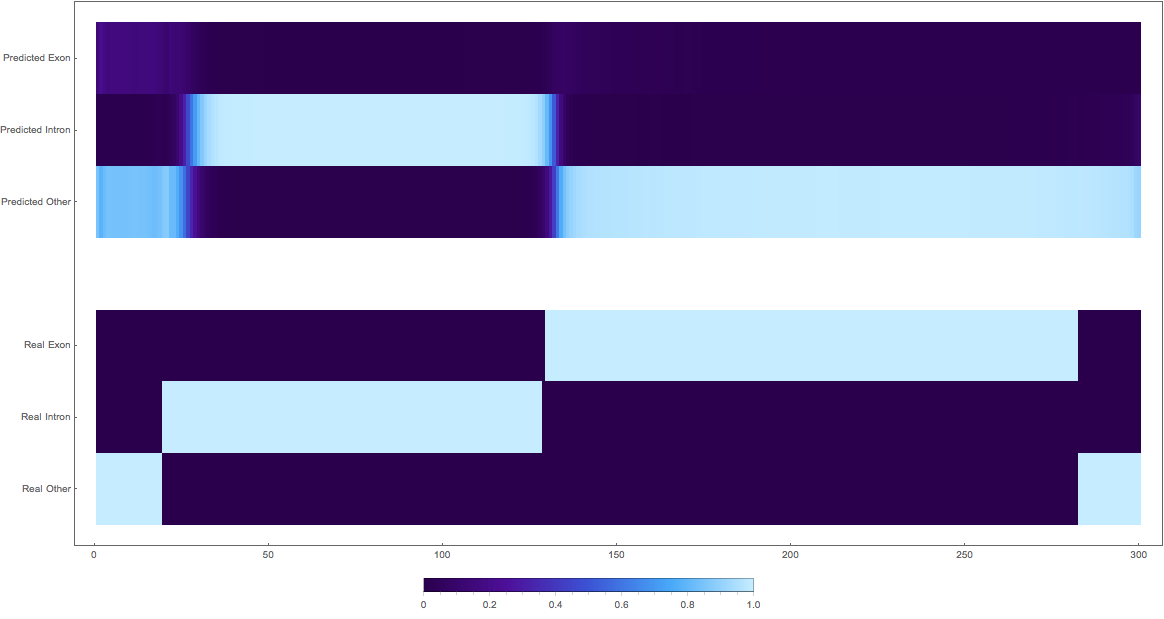
2. (reversed and wrong class)
3. (maybe this is an exon?) 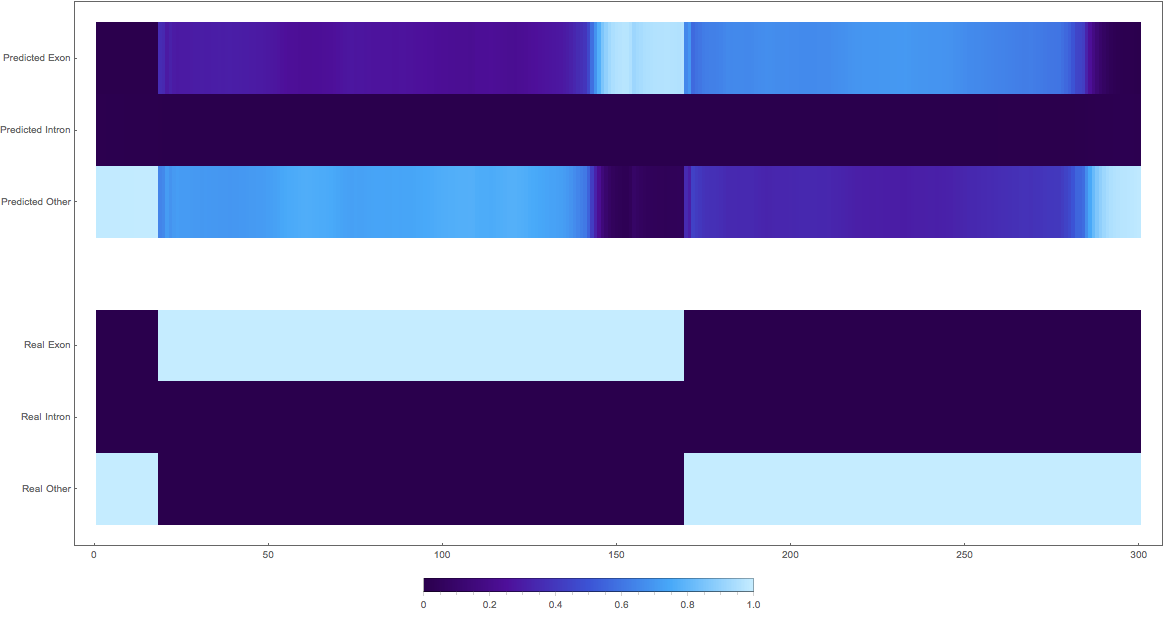
The confusing
1: very pretty
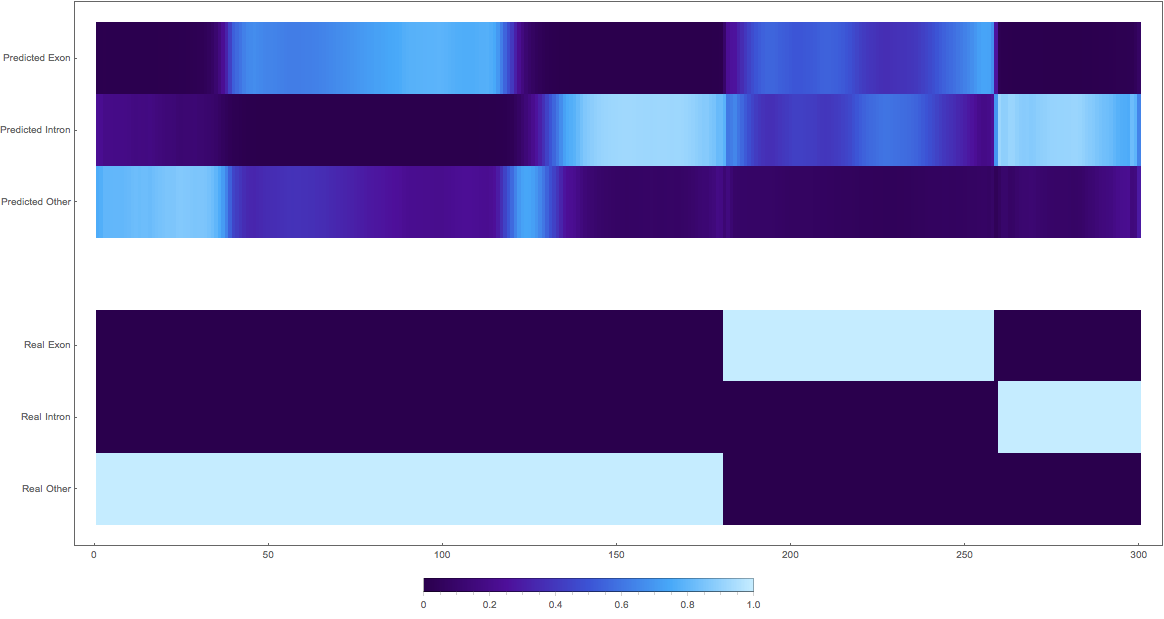
2: shifted 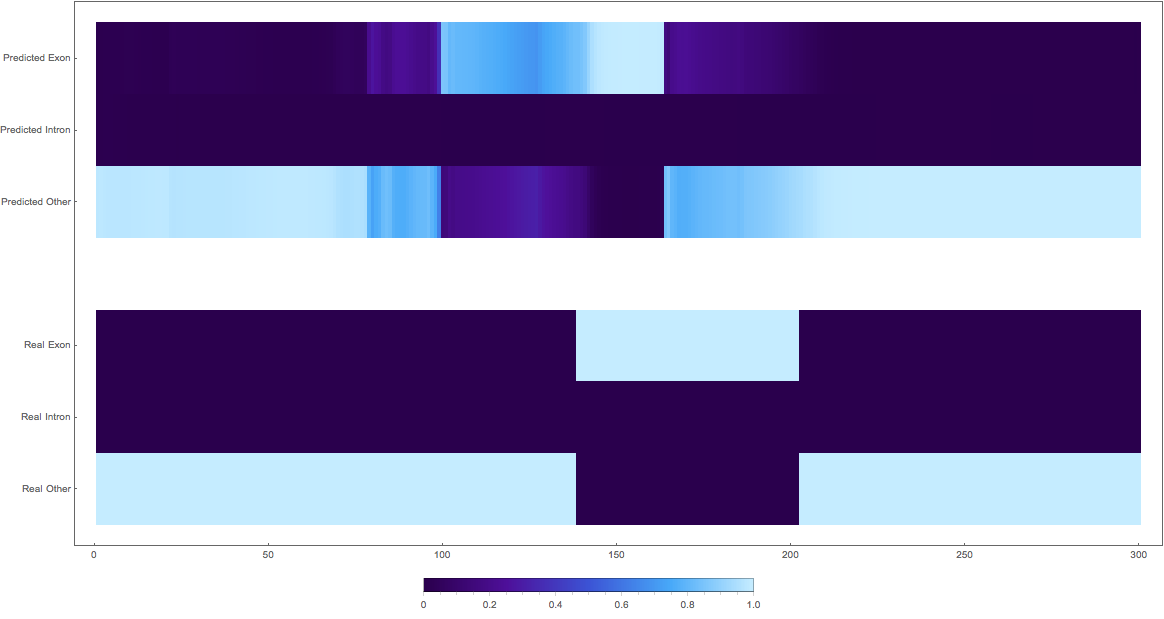
3: shifted wrong classes 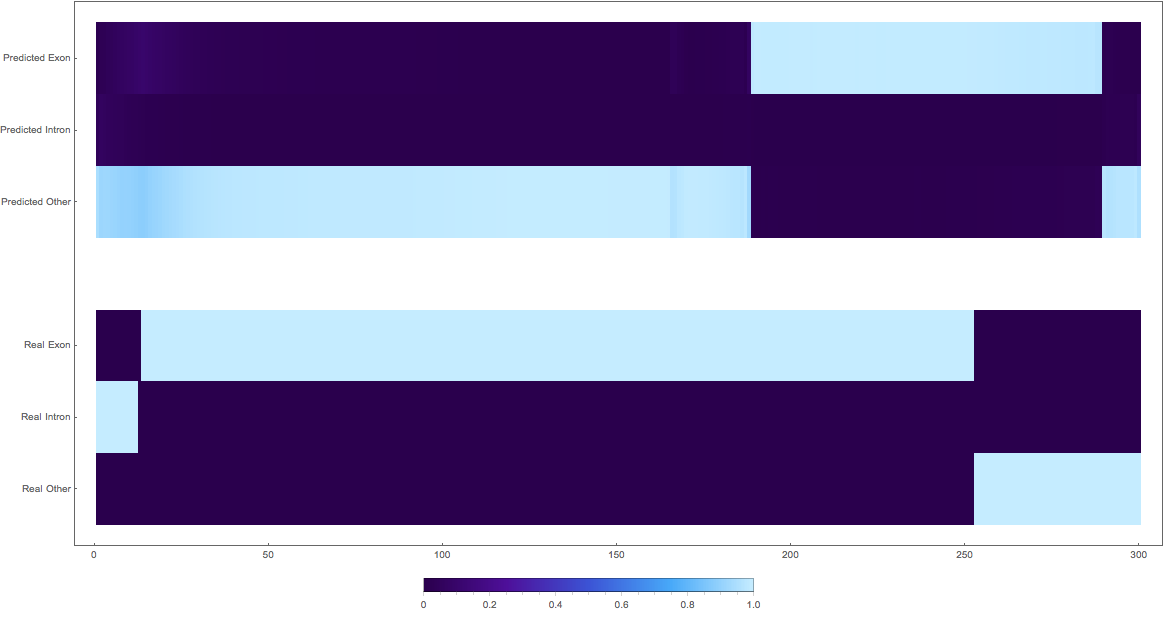
Squared Loss Per Pixel
Discussion and future direction
Semantic segmentation with inception architecture works much better than other instance detection networks. I am considering trying Mask-RCNN.
Input as to other approaches for solving this problem welcome.


