The past summer I attended the Wolfram Science Summer School and worked with machine learning to infer evolutionary relationships (I used images of ants). Later I was offered to continue this project as part of the Wolfram Mentorships Program. For this project we decided to focus on sequences of biological data, which typically are genes or proteins, which is what modern phylogenetics use.
Brief introduction to Phylogenetics:
Phylogenetics is an area of biology which focuses on revealing the evolutionary relationships between biological entities, which can be taxa (groups of organisms) or even genes or proteins. Modern phylogenetic studies use sequences of morphological or genetic data, and use the idea that all these sequences evolved from a common ancestor, so the difference between sequences is related to the evolutionary distance between them, that is, the farther in the past two organisms shared a common ancestor, the more differences their sequences will have. Discovering these evolutionary relationships is a problem of hierarchical clustering, more on phylogenetics can be found in Wikipedia: https://en.wikipedia.org/wiki/Phylogenetics.
Using WL to infer a phylogenetic tree with biological data:
For this project I downloaded a set of sequences used in the study "Phylogenomics Resolves Evolutionary Relationships among Ants, Bees, and Wasps" by Johnson, Briar R. et al. Current Biology, V. 23, Issue 20, 2058 - 2062. This was to have a phylogenetic tree to compare to. The data set had 19 amino acid sequences, each more than 175'000 characters long, previously aligned. An example of a fragment is: "??????MLRFAARATSA---RQS", where the character "?" represents an unknown amino acid, "-" represents a gap, and the letters represent an amino acid residue. The amino acid residues and gaps are the most informative when used to construct a phylogenetic tree.
The data was downloaded from http://datadryad.org/resource/doi:10.5061/dryad.jt440, where the authors of the study deposited it. With some manual cleaning, like changing the names of the species in the files (some were written in an abbreviated form or referred to the name of the subfamily), i had an association where each value was around 175,000 characters long. When I mention a "site" I refer to a position in all sequences. To remove sites where there was at least 1 unknown amino acid I used the following code:
(* The association with the original data is specieAminoAcidAssoc,
and as it is very long I did not add that to this post. *)
noUnknowns =
StringJoin[#] & /@
Transpose[
Cases[Transpose[Characters[Values[specieAminoAcidAssoc]]],
x_ /; Count[x, "?"] == 0]];
Using the full dataset would take too much time, so I first I removed all sites where any sequence had an unknown amino acid residue ("?"), keeping a total of 43,833 sites. Then I selected 5,000 random sites, as using the 43,833 sites took to long. Testing with different sizes less than 5,000 showed little change, which leads me to believe that using 10,000 sites will not be very different than using all 43,833.
(* A subset of small data, made of random sites. generaNames is a list written
by me to replace the names that each sequence had, as the original names were
sometimes abreviations or referred to the subfamily of the sample. *)
generaNames = {
"Apis", "Argochrysis", "Brachycistis", "Bradynobaenidae", "Bombus",
"Chyphotes", "Crioscolia", "Harpegnathos", "Lasioglossum",
"Linepithema", "Megachile", "Mischocyttarus", "Nasonia",
"Pogonomyrmex", "Pepsis", "Pseudomasaris", "Sceliphron",
"Sphaeropthalma", "Stigmatomma"
};
randomSample =
StringJoin /@
Transpose[
RandomSample[
Cases[Transpose[Characters[noUnknowns]],
x_ /; Count[x, "?"] == 0], 5000]];
randomSampleAssoc = <||>;
Do[randomSampleAssoc[generaNames[[i]]] = random[[i]], {i,
19}]
The data was saved to the file clusteringData.m in the working directory, to run this you may have to save the .m file to your directory.
data = << clusteringData.m;
(*Using ClusteringTree on the data *)
tree = ClusteringTree[data]
The following tree has its labels colored to compare it to the tree the original study inferred.

The tree obtained by the authors of the study is:
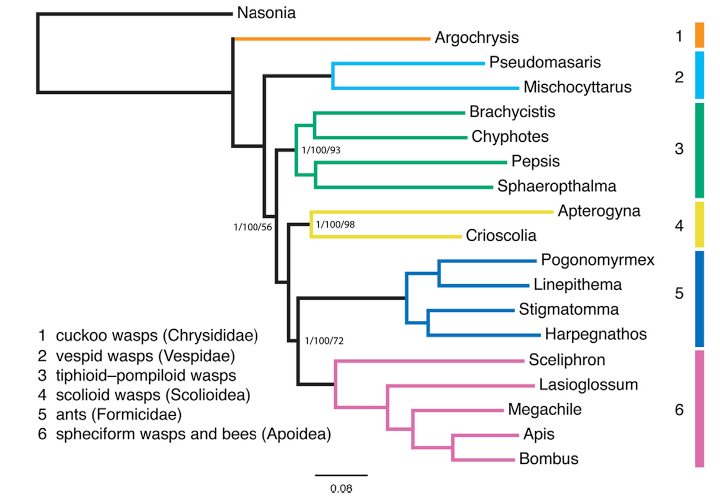
When the tree obtained with ClusteringTree is compared to this tree, it is important to notice that two major lineages, Formicidae (blue), Apoidea (purple) and Vespidae (cyan) are represented in both trees. Nasonia,the outgroup is almost the most separated group in the tree made with ClusteringTree. Other similarities and differences can be found by observing both trees.
Final Thoughts
The methods used by the function ClusteringTree and Dendrogram have been taken into account when developing algorithms that infer phylogenies from biological data, and many modern studies on phylogenetics use more complex algorithms, usually based on Bayesian inference. This project was made to show that with the Wolfram Language it is possible to study the evolutionary relationships of different organisms, and it would be interesting to see how more sophisticated machine learning algorithms could do with these problems.
I would like to thank Alison Kimball and Todd Rowland for their support and guidance in this project.
Update
I just added the files that were missing (below) and the code used to clean the data. I began explaining that the original data was in the association specieAminoAcidAssoc instead of explaining how I made that association because it involved only importing the file and removing blank lines and stuff like that, which I thought took attention away from the important code.
 Attachments:
Attachments:


