Background:
Viruses have been classified in the taxonomy tree using by unifying molecular and morphological data. However, viruses within the same genus might vary greatly in their potential to infect human beings. A simple example is the Flavivirus genus which contains species such as Zika, Dengue, West Nile and Yellow Fever viruses, all of which are pathogenic to humans. At the same time, the genus also contains species such as the Palm Creek virus and Tembusu virus(causes infection in ducks) that have been shown to be harmless in humans.
The goal of the project here is to train Convolutional Neural Networks to identify pathogenic viral species from their corresponding genomic sequences.
Compiling Data
Viral genomic sequences can be downloaded via FTP from the NCBI Reference Sequence Database(RefSeq) in the FASTA format. This FASTA header is of the format,
> ref|AccessionID|Species Name, complete genome
Example,
> ref|NC_021865.1| Paenibacillus phage phiIBB_Pl23, complete genome
In order to label viral genomes as 'pathogenic' and 'non pathogenic', annotations can be extracted from the Human Disease Ontology. The ontology can be hosted locally as a Neo4j instance and queried for viral infectious agents. Unfortunately, the ontology contains a mapping of diseases to taxonomy ids of the causative species. NCBI Taxonomy provides a mapping of the accession ID to taxonomy ID. This file was downloaded and the accession IDs were mapped to the Taxonomy IDs.
This assembled dataset is hosted publicly for use on the Wolfram Data Repository and is available here along with the commands required to use the resource.
Dinucleotide usage has been shown to differentiate between viruses that affect different hosts in Flaviviruses. This compiled dataset can be used to create a quick PCA plot of the Dinucleotide usage of all 9334 viral genomes to see if there is indeed a correlation across other species as well.
ds = ResourceData["749327c2-fbd2-42db-b39c-597a7773057c"];
cscheme = Normal@ds[All, "Pathogen"] /. {
"False" -> Directive[Opacity[.15], Blue] ,
"True" -> Directive[Red, PointSize[Large]]
};
pcaPoints =
{
Pick[dinPCA[[All, 1 ;; 2]],
Not /@ ToExpression@Normal@ds[All, "Pathogen"]
],
Pick[dinPCA[[All, 1 ;; 2]],
ToExpression@Normal@ds[All, "Pathogen"]
]
};
ListPlot[pcaPoints,
PlotStyle -> {
Directive[Opacity[.35], Blue],
Directive[Red, PointSize[0.012]]
},
AxesLabel -> {"PC1", "PC2"},
PlotLabel -> "PCA of dinucleotide usage",
PlotLegends -> {"Non Pathogenic", "Pathogenic"}
]
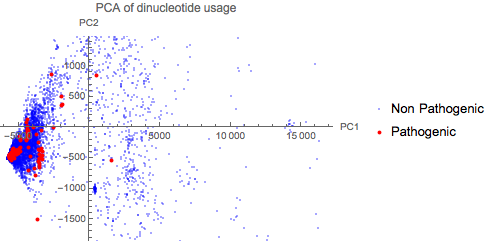
The plot above shows that there is no obvious correlation between dinucleotide usage and pathogenicity.
Preprocessing data to get Training and Test datasets
A convolutional neural network is good at identifying patterns and achieves very high accuracy is tasks such as image processing. The goal here is to see if such a network can identify features to distinguish between 'pathogenic' and 'non pathogenic' viral genomes. Convolutional neural networks accept a fixed length of input. In order to facilitate this, the genomes are divided into chunks of 1000 bps each with chunks smaller than that being discarded.
To create a relatively balanced training and test set, genomes of lengths between 25000 and 2000 are chosen from both the 'pathogenic' and 'non pathogenic' groups.
pathogenic = ds[Select[#Pathogen == "True" &], "Genome"];
pathogenic =
Select[pathogenic,
StringLength[#] <= 25000 && StringLength[#] >= 2000 &];
nonpathogenic = ds[Select[#Pathogen == "False" &]];
nonpathogenic =
Take[Select[
nonpathogenic, (
StringLength[#Genome] <= 25000 &&
StringLength[#Genome] >= 2000) &], 144];
A function is written to chunk the genomes into sizes of 1000 bps.
chunkGenome[x_, k_] :=
Normal@Table[
Table[StringTake[x[[g]], {i, i + k - 1}], {i, 1,
StringLength[x[[g]]] - k, k}], {g, 1, Length[x]}]
This function is then applied to both the pathogenic and non pathogenic groups of sequences,
chunksPathogenic = chunkGenome[Flatten[Normal /@ {pathogenic}], 1000];
chunksNonPathogenic =
chunkGenome[
Flatten[Normal /@ {shortestNonPathogenic[All, "Genome"]}], 1000];
Sometime when viral genomes are sequenced, some positions of nucleotides are ambiguously labelled because of the lack of confidence in the quality of sequencing at that point. Such sequences are removed to ensure that bases labelled as 'R' etc are not present in the training and testing datasets.
chunksPathogenic =
Select[Flatten[
chunksPathogenic], (StringDelete["A"]@
StringDelete["T"]@StringDelete["G"]@StringDelete["C"]@#) ===
"" &];
chunksNonPathogenic =
Select[Flatten[
chunksNonPathogenic], (StringDelete["A"]@
StringDelete["T"]@StringDelete["G"]@StringDelete["C"]@#) ===
"" &];
Now, the first 500 of these sequences are used for training and the rest for testing.
trainingset =
RandomSample[
Join[Thread[Normal@Take[chunksPathogenic, 500] -> "Pathogenic"],
Thread[Normal@Take[chunksNonPathogenic, 500] ->
"NonPathogenic"]]];
testset =
RandomSample[
Join[Thread[
Normal@Take[chunksPathogenic, { 501, 556}] -> "Pathogenic"],
Thread[Normal@Take[chunksNonPathogenic, { 501, 556}] ->
"NonPathogenic"]]];
The Neural Network
A convolutional neural network with the configuration shown below was trained on the training set. The string input was hot encoded into a vector and then transposed before the convolution operations in the neural network.
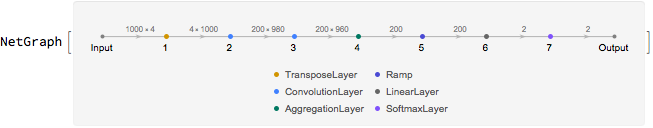
The neural network achieves an accuracy of 76% on the test set.
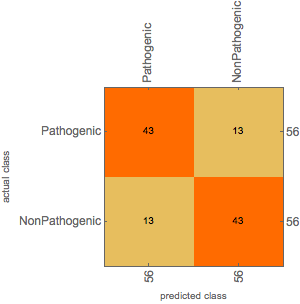
The Confusion matrix on the training set is shown below,
Future Direction
Expanding the size of the training and test set will help reduce overtraining of the network. The configuration of the neural network has to be tweaked further to attain higher accuracy. Hybrid neural networks with both convolution and recurrent neural layers seem to hold a lot of promise[1]. On the preprocessing end, instead of chunking down the sequences into 1000 bps, they can be converted into a sparse genome-feature[4].
Further Reading and References
- DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences
- Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks
- West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses
- k-mer Sparse matrix model for genetic sequence and its applications in sequence comparison


