ABSTRACT
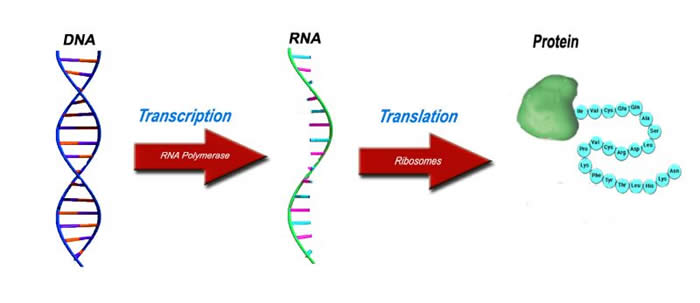
A living cell has a vast amount of enzymes and molecules that have influence on the derivation of a protein from a gene. Protein synthesis is one of the most complex processes in the cell and it involves a lot of signals, enzymes, and other molecular complexes. As the central dogma of molecular biology says, our DNA undergoes processes that make it into an RNA sequence (Transcription), and the latter to an amino acid sequence (Translation); amino acids then fold into proteins. In this pipeline, two essential enzymes are the spliceosome and the ribosome. Spliceosome cuts off the unnecessary parts (Introns) from the primary RNA transcript and joins the parts (Exons) which then have to be translated into an amino acid sequence. This produces a mature mRNA. The ribosome, in its turn, takes the mature mRNA and translates it into its corresponding amino acid sequence.
The aim of this project is to model the primary RNA transcript ?mRNA?Amino Acid Sequence pipeline. We used Machine Learning techniques to design and train an Artificial Neural Network to recognize all the important sites that take part in these processes and imitate some of the algorithms that the cell does during protein synthesis.
I. Exon-Intron Splicing
Data Acquisition
In the first step, we import all of the data about the special sites that will be needed. Data on Intron and Exon splice site regions were acquired from a Machine Learning Database.
Firstly, we import around 3160 sequences from the database. The sequences are labeled "EI" for an Exon-Intron splice site, "IE" for an Intron-Exon splice site and "N" for Neither. Throughout the project, we converted the sequences to 2-dimensional vectors by converting the nucleotide letters A, C, G and T to {1,0,0,0}, {0,1,0,0}, {0,0,1,0} and {0,0,0,1}, respectively. In this way it is easier for the classifier to understand the data.
Classifier
We make an association of sequences and their labels and Classify this data.

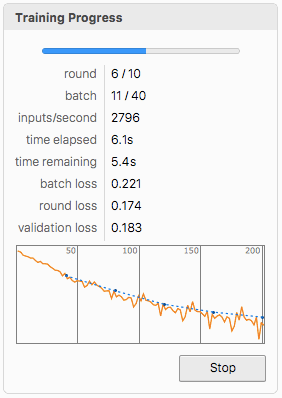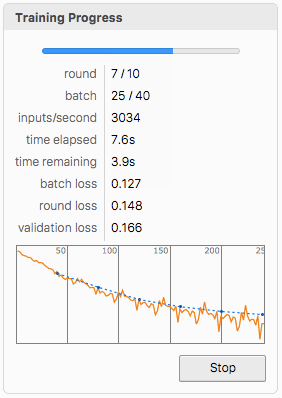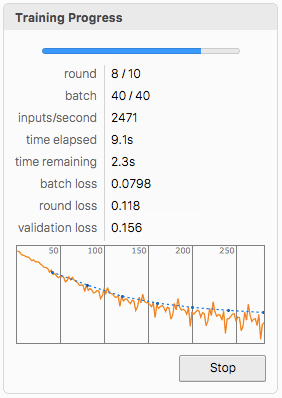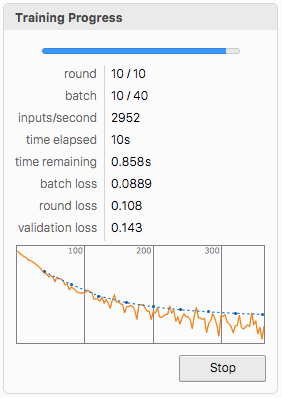
Artificial Neural Network Design and Training (Optional)
We also propose an ANN that is trained on the acquired data as an alternative for the classification of splice sites. The user needs to format the data into grayscale images for dimensions {60, 4} to test the network. Formatting of the data is also presented below. (Important: The whole dataset has been used for training, so the user needs another one.)
Sliding Window
To analyze the sequence and to find splice sites in it, we construct a 60 nucleotides long "window" that slides along the primary RNA transcript and looks for splice sites.
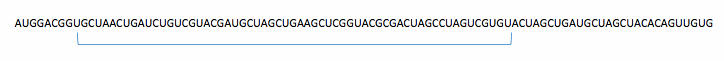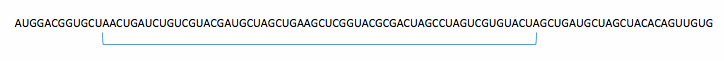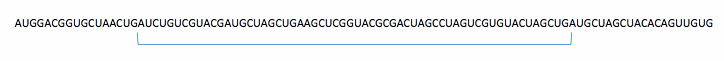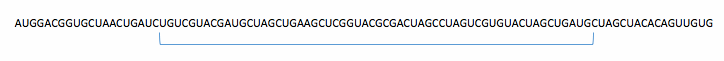
takeIdxSeq[seq_, idx_] := If[idx <= Length@seq, Take[seq, {idx, 59 + idx}]]
scoreEI[str_String] :=
Module[
{p, k, l, seq, splicesites, out},
seq = Characters[str] /. {"A" -> {1, 0, 0, 0}, "C" -> {0, 1, 0, 0}, "G" -> {0, 0, 1, 0}, "T" -> {0, 0, 0, 1}};
splicesites = ssList[str];
k = Take[seq, {#, # + 59}] & /@Flatten@Position[splicesites, "EI"];
l = takeEIRange[#] & /@ k;
p = findPositions[#] & /@ l;
Thread[{score[pwmDon, Part[p, #]] & /@ Range[Length@p], "EI", Flatten@Position[splicesites, "EI"]}]
]
scoreIE[str_String] :=
Module[
{p, k, l, seq, splicesites, out},
seq = Characters[str] /. {"A" -> {1, 0, 0, 0}, "C" -> {0, 1, 0, 0}, "G" -> {0, 0, 1, 0}, "T" -> {0, 0, 0, 1}};
splicesites = ssList[seq];
k = Take[seq, {#, # + 59}] & /@
Flatten@Position[splicesites, "IE"];
l = takeIERange[#] & /@ k;
p = findPositions[#] & /@ l;
Thread[{score[pwmAcc,Part[p, #]] & /@ Range[Length@p],"IE", Flatten@Position[splicesites,"IE"]}]
]
Donor Site and Acceptor Site PWMs
The Position Weight Matrices (PWMs) are commonly used to represent motifs (patterns) in biological sequences. These two are acquired from the same sequences from the database.
- Donor Site (Exon-Intron) PWM

- Acceptor Site (Intron-Exon) PWM

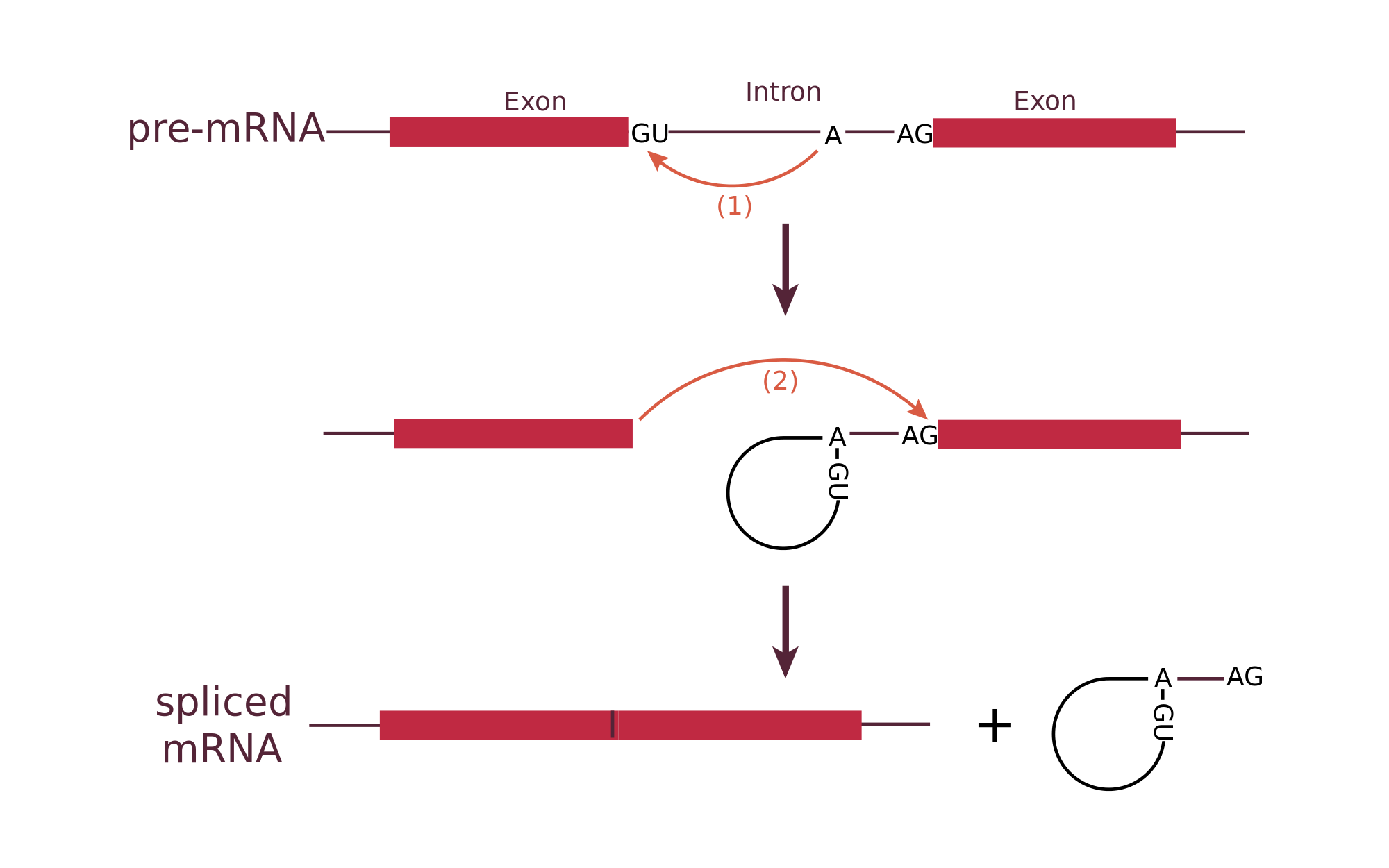
Removing the wrong Splice Sites
This is one of the most crucial steps in the whole processes. An mRNA has to start and end with Exons, so if there is a IE splice site near the beginning or a EI splice site near the end of the sequence, this means that the splice sites need to be further filtered.
findMaxpos[list_, idx_] := Flatten@Position[list[[All, 1]], Max[list[[All, 1]]]] + idx - 1;
FindWrongSS[seq_String] :=
Module[
{listEI, listIE, list, eipos, iepos, m1, m2, k1, k2, j1, j2, f1, f2, v1, v2, out},
listEI = scoreEI[seq];
listIE = scoreIE[seq];
list = SortBy[Partition[Flatten@Append[listEI, listIE], 3], Last];
eipos = Flatten@Position[list[[All, 2]], "EI"];
iepos = Flatten@Position[list[[All, 2]], "IE"];
m1 = {Min[#], Max[#]} & /@ Split[eipos, #2 - #1 == 1 &];
m2 = {Min[#], Max[#]} & /@ Split[iepos, #2 - #1 == 1 &];
k1 = DeleteCases[m1, a_ /; First[a] == Last[a]];
k2 = DeleteCases[m2, a_ /; First[a] == Last[a]];
j1 = Flatten[findMaxpos[Part[list, First@# ;; Last@#], First@#] & /@ k1];
j2 = Flatten[findMaxpos[Part[list, First@# ;; Last@#], First@#] & /@ k2];
f1 = Flatten[Range[First@#, Last@#] & /@ k1];
f2 = Flatten[Range[First@#, Last@#] & /@ k2];
v1 = list[[#, 3]] & /@ Complement[f1, j1];
v2 = list[[#, 3]] & /@ Complement[f2, j2];
out = Flatten[Append[v1, v2]];
out
]
removeUnnecessary[seq_String] :=
Module[
{list, firstEI, firstIE, lastEI, lastIE, splicesites},
list = FindWrongSS[seq];
splicesites = ssList[seq];
splicesites[[list]] = "N";
firstIE = Part[First@Position[splicesites, "IE"], 1];
firstEI = Part[First@Position[splicesites, "EI"], 1];
lastIE = Part[Last@Position[splicesites, "IE"], 1];
lastEI = Part[Last@Position[splicesites, "EI"], 1];
If[firstIE < firstEI, splicesites[[firstEI]] = "N"];
If[lastIE < lastEI, splicesites[[lastEI]] = "N"];
splicesites
]
Final Step: Splicing Introns and Joining Exons
Spliceosome[seq_String] :=
Module[
{a, b, k, m, positions, str},
a = Flatten@Position[removeUnnecessary[seq], "EI"];
b = Flatten@Position[removeUnnecessary[seq], "IE"];
k = Partition[Sort@Flatten@Append[a, b], 2] + 30;
m = Flatten[Range[First@#, Last@#] & /@ k];
positions = Partition[Complement[Range[Length@seq], m], 1];
str = Delete[Characters[seq], Partition[m, 1]];
StringJoin[str]
]
II. mRNA?Amino Acid Translation
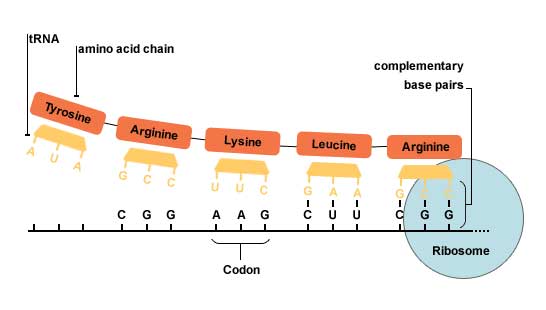
Nucleotides (A,C,G,U) make triplets called codons. 61 of the 64 possible triplets code for amino acids (one codes for Met, which is the encoded by the codon AUG and in most of the cases that is the start codon), while the other 3 are stop codons, which stop the translation.
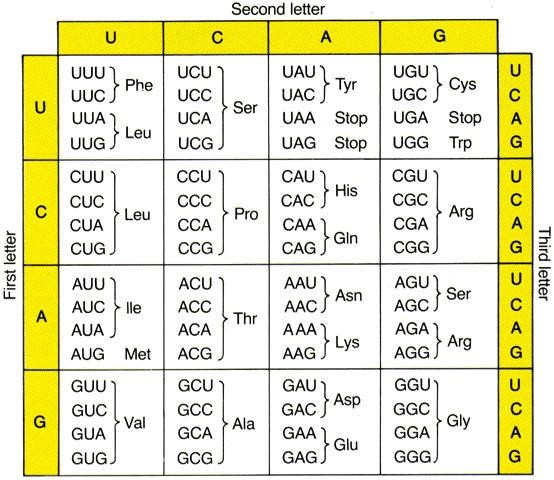
Ribosome[seq_String] :=
Module[
{findStartCodon, concatinateSeq, translate, findStopCodon, sp, codonlist, prot, out},
findStartCodon[seq2_String] :=First@Flatten@StringPosition[seq2, "AUG"];
concatinateSeq[n_Integer, seq1_String] := StringPartition[StringTake[seq1, {n, -1}], 3];
translate[codons_List] := CodontoAminoAcid[#] & /@ codons;
findStopCodon[prot_List] := Take[prot, {1, First[Flatten@Position[prot, "*"]]}];
sp = findStartCodon[seq];
codonlist = concatinateSeq[sp, seq];
prot = translate[codonlist];
out = findStopCodon[prot];
out
]
Final Step: Primary RNA ? Amino - Acid
Wrapping up all of the processes ?
RNAtoAA[seq_String] :=
Module[
{spliced},
spliced = Spliceosome[seq];
Ribosome[spliced]
]


