The idea I would like to share comes from the domain of biological sciences. Turing patterns are widespread in the biological and the physical world. Named after Alan Turing (for his 1952 paper, the chemical basis of morphogenesis), Turing patterns in biology (think about pretty Zebra strips) involve two classes of species: activators and inhibitors. The activators are called so because they can trigger their own production as well as in some cases produce their antagonists - the inhibitors. On the other hand, the inhibitors are called so because their purpose is to hinder the activity of activators. These activators and inhibitors come in different flavours. Nevertheless, the point to remember is that both activators and inhibitors can form spatial gradients by virtue of reaction/diffusion in a medium, and it is these gradients that in turn are responsible for generating patterns - for example the case of Zebra strips.
Now one may ask the question: how do inhibitors inhibit and activators carry out their jobs?
Actually, there is a third specie at play too: the membrane molecules. These particular class of molecules are present on the surface of biological cells. These membrane bound molecules are termed as receptors (for their ability to receive other molecules - the ligands). The freely diffusing molecules, for instance, activators and inhibitors can interact with the receptors. When an activator ligand binds with the receptor it triggers a cascade of events that lead to the its own production. Moreover, as aforementioned, the same cascade may generate inhibitors. Now, when an inhibitor interacts with the receptor it ensures that the activators have one less receptor to bind to. Therefore, inhibitors serve to block the interaction of membrane receptors and activators. Having said that, there may be other mechanisms via which inhibitors can interfere with an activator's activity. Nevertheless, the system that will be under discussion here deals with inhibitors that solely interfere by merely blocking the activator from binding with the receptor.
In the nodal/lefty system, the nodal molecules are the activators whereas the lefty is the inhibitor. Both these ligand molecules compete for the same receptor. The rule of the game is that a nodal binding event triggers its own production as well as the production of lefty. A lefty binding event blocks nodal from accessing receptors. Since biological molecules have an affinity to degrade, therefore, the system has a tendency to probabilistically eliminate molecules.
In my simulation what I initially do is to create a bunch of nodal (blue species) and lefty (red species) inside the cell and receptors on the membrane of a cell (to a first approximation modeled a circle). The system could have infinite configurations to begin with so one such configuration is depicted below:
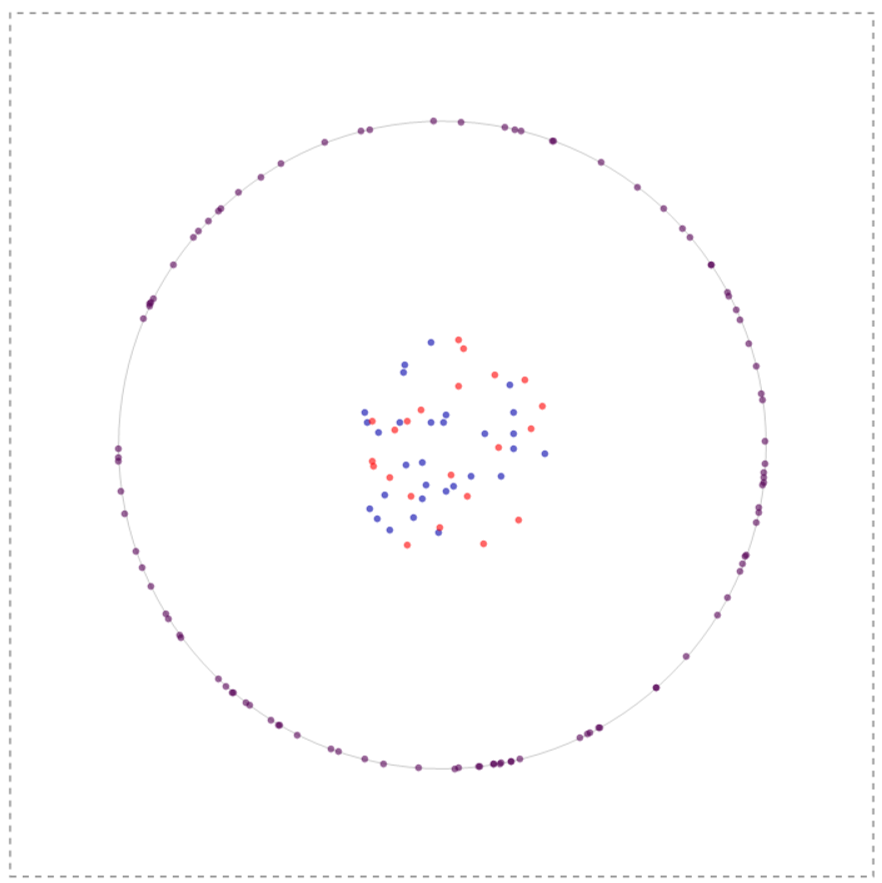
All these molecules have the ability to diffuse using a gaussian distribution as a stepping function with the width of the gaussian defined by 2 Sqrt[D t] where D is the Diffusion coefficient and t is the time-step of observation. The ligands (nodal and lefty) are crudely modeled as being produced inside the cell but with a tendency to move outside the cell (by virtue of Brownian motion); once outside they cannot enter the cell, nevertheless can interact with the receptor molecules on the membrane or roam within the box aimlessly. Furthermore, reiterating, the ligands can degrade and have the ability to inhibit or activate depending upon which ligand interacts with the receptor.
If one runs the simulation, in time the system evolves spatially as can be inferred from the image below: 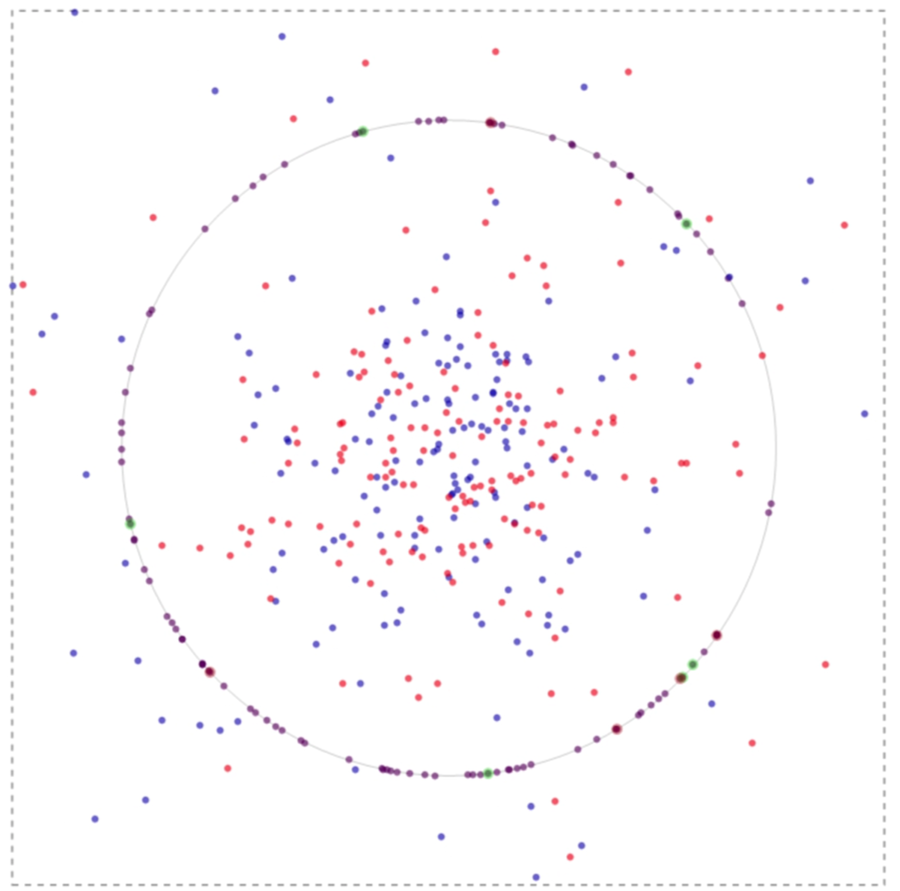
The green spots on the membrane in the image above implicate to the events where the binding is associated with nodal molecules and receptors. Likewise, a red spot points to a lefty molecule binding to a receptor.
To simulate the system, I used the Wolfram Language as the language of choice owing to its powerful arsenal of built-in in functions, and its ability to perform complex tasks with ease (thanks to the rule-based and pattern matching schemes). Each of the helper functions has a particular task: for instance to ensure diffusion of free ligands and receptors, to bar the ligands from entering the cells, using a Nearest Function approach to determine interactions between the free ligands and the free receptors (receptors that are available for binding), a function to facilitate the binidng event, to degrade ligands and another to produce ligands, so on and so forth. Something to bear in mind is that not every interaction is successful (game of chance) and the ligand (irrespective whether nodal or lefty) can separate from the receptors if bound (also defined probabilistically).
I will present the code for simulating such a system below in a stepwise manner:
We can create the bounding regions to confine the molecules
\[ScriptCapitalR] =
ImplicitRegion[
x^2 + y^2 <= 0.25^2, {x,
y}]; (* defining the region containing the molecules in a single \
cell *)
(* creating necessary regions and constraints *)
Subscript[\[ScriptCapitalR], 2] =
ImplicitRegion[
x^2 + y^2 <= 0.75^2, {x, y}]; (* creating the circular cell *)
Subscript[\[ScriptCapitalR], 3] =
BoundaryMeshRegion[{{-1, -1}, {1, -1}, {1, 1}, {-1, 1}},
Line[{1, 2, 3, 4, 1}]]; (* creating the bounding box *)
The following code can be used to create initial configuration of particles:
(* setting up initial configuration of ligands *)
initialParticleConfig[name_String, {x_?NumberQ, y_?NumberQ},
number_?IntegerQ] :=
Module[{specie, specieBoundaryIndex, speciePts},
specie = RandomReal[{x, y}, {number, 2}]; (*
this creates a number of particles for specified domains *)
specie = Select[specie, # \[Element] \[ScriptCapitalR] &]; (*
select those particles that are present within the cell *)
specie =
Association@
MapIndexed[First@#2 -> #1 &, Thread[{name, specie}]]; (*
lets thread the particles with a rule where the particles are \
associated with an index, their name and their coordinates *)
specieBoundaryIndex = (#[[2]] \[Element] \[ScriptCapitalR]) & /@
Values[specie] /. {True -> 0, False -> 1}; (*
a mapping over particle position to check whether the particles \
are outside the defined boundaries (cells) or within it.
a particle inside would yield True (replace True with 0) and \
external to the region will yield False (replace with 1) *)
specieBoundaryIndex =
Association@Thread[Range[Length@specie] -> specieBoundaryIndex]; (*
putting an index to the previous result for unique identification \
of particle *)
speciePts = Values@specie[[All, 2]]; (*
this will only extract the particle positions *)
Return@{specie, specieBoundaryIndex, speciePts};(*
now i can return three variables. specie (<|
particle number \[Rule] {{"Nodal || Lefty"},{x,y}} ... |> ),
specieBoundaryIndex (<| particle number \[Rule] 1 ||
0 ... |>) and speciePts {{x,y}....} *)
];
(* setting up configuration of membrane proteins *)
membraneReceptorConfig[{x_?NumberQ, y_?NumberQ}, radius_?NumberQ,
number_?IntegerQ] := Module[{membranePts, membraneIndex},
(* this creates a random number of particle on a circle,
basically representing membrane proteins *)
membranePts = Table[RandomPoint[Circle[{x, y}, radius]], number];
membraneIndex =
Thread[Range[number] ->
Thread[{membranePts, 0, None, "type"}]]; (*
putting an identification number for the previous result i.e.
receptor number \[Rule] {coord, 0 or 1, BoundTo/None, Type-
Ligand} *)
Return@{membranePts, membraneIndex} ;(* returns membranePts ({x,
y}....) and membraneIndex (membrane protein number \[Rule] {x,y},
0,None,Type) *);
];
Helper functions to perform various activities:
the functions below allow the ligands to diffuse
BrownianWalk[number_, elem_] :=
Module[{oldposition = elem[[2]], newposition, step, tag = elem[[1]]},
step = {RandomVariate[NormalDistribution[0, 0.01]],
RandomVariate[NormalDistribution[0, 0.01]]}; (*
take a random number from a Gaussian where the width can be defined \
as 2 Sqrt[Subscript[D, coeff] \[Delta]t] *)
newposition = oldposition + step; (*
add the new step to the original particle position *)
number -> {tag, newposition} (*
thread the particle with its type and new position *)
]
BrownianFunc[assoc_] := Module[{temp},
temp = Association[KeyValueMap[BrownianWalk, assoc]]
]
for making the membrane receptors to diffuse but confined to the membrane
(* strategy: get all the membrane pts. rotate them using a random number (angle) generated from a Gaussain function. export
updated membrane pts out of the module *)
BrownianWalkMembrane[receptorpos_, membraneind_] :=
Module[{membranepos, membrane, angle, center},
center = {0, 0};
angle = \[Pi]/1800.; (* 0.1 degree rotation about center {0,0} *)
membranepos =
RotationTransform[RandomVariate[NormalDistribution[0, angle]],
center][#] & /@ receptorpos; (*
here we generate the new position *)
membrane =
Table[i -> {membranepos[[i]],
Sequence @@ membraneind[[i, 2, 2 ;;]]}, {i, 1,
Length@membraneind}]; (*
updating the position in the membraneindex *)
{membranepos, membrane} (*
exporting the membrane position and the updated membraneindex *)
] /; Length@membraneind > 0
to ensure the ligands do not leave the system or enter cell once outside
ligandCheck[particles_, speciecopy_, particleBoundaryIndex_] :=
Module[{specie = particles, speciekeys, coordsspecie,
indexspecie, threadList},
speciekeys = Keys@specie; (* the indices of the particles *)
coordsspecie = Values@specie[[All, 2]]; (*
the positions of the particles *)
indexspecie = Values@particleBoundaryIndex; (*
whether particles are present inside 0 or outside the cells 1 *)
threadList = Thread[{speciekeys, indexspecie, coordsspecie}]; (*
lets thread the above three together *)
threadList =
threadList /. {ind_?IntegerQ, x_?IntegerQ, y_List} /;
x == 0 && !
y \[Element] Subscript[\[ScriptCapitalR], 2] :> {ind, 1,
y}; (* update a particle that was inside but takes a step \
outside *)
threadList =
threadList /. {ind_?IntegerQ, x_?IntegerQ, y_List} /;
x == 1 && y \[Element] Subscript[\[ScriptCapitalR], 2] :> {ind,
1, speciecopy[ind][[2]]}; (*
prevent particles that are outside to step inside *)
threadList =
threadList /. {ind_?IntegerQ, x_?IntegerQ, y_List} /;
x == 1 && !
y \[Element] Subscript[\[ScriptCapitalR], 3] :> {ind, 1, speciecopy[ind][[2]]};
(* prevent particles to leave the bounding box *)
threadList (* final check goes outside the module without using Return *)
]
determine interactions and create binding events for successful interactions
nearestFunction[ligandList_, membraneIndex_, membranepts_] :=
Module[{freeReceptors, receptormap, interactions, ligandlist},
ligandlist = Cases[ligandList, {ind_, 1, pos_} :> {ind, pos}]; (* lets find particle index and positons for ligands
outside the cell *)
freeReceptors =
Cases[membraneIndex,
PatternSequence[x_ -> {y_List, 0, None, "type"}] :> y -> x] ; (* determine free receptors and invert the index and positions
for making nearest function *)
receptormap = Nearest[freeReceptors]; (* creates a nearest function from the freereceptors *)
interactions =
Reap[Map[Function[{particle},
Sow[particle[[1]], receptormap[particle[[2]], {1, 0.01}]]],
ligandlist], _, List][[2]] //
DeleteDuplicatesBy[
Replace[#, {x_Integer, {y_Integer, z___Integer}} :> {x, y}, 2],
Last] &; (* this finds the nearest receptor to a ligand within a defined radius. since many ligands maybe close to a
receptor we can use replace to limit to the interaction to a single receptor-ligand pair for all possible receptors *)
Reverse[interactions, 2] (* reversing the {{receptor, ligand}..} pairs and exporting from module *)
] /; Count[membraneIndex, 0, 3] > 0 && Length@ligandList > 0 (* run only if free receptors as well as free ligands present *)
biasSharedReceptors[nod_, left_] :=
Module[{nodal = nod, lefty = left, union, probability},
union = Cases[GatherBy[Join[nodal, lefty], Last], {_, x_} ..];
Map[
Function[{part},
probability = RandomReal[];
If[probability > 0.5,
lefty =
Delete[#, Flatten@Position[#, Flatten@Intersection[part, #]]] &@lefty,
nodal =
Delete[#, Flatten@Position[#, Flatten@Intersection[part, #]]] &@nodal]
], union];
Return@{nodal, lefty};
] /; (nod =!= {} && left =!= {}) (* If a receptor is interacting with both nodal and lefty at the same time,
bias the system to choose one *)
bindingEvent[membraneIndex_, {ligandpos_, receptorpos_}, ligandnom_] :=
Module[{coord},
coord = membraneIndex[[receptorpos]][[2, 1]];
ReplacePart[membraneIndex,
receptorpos -> (receptorpos -> {coord, 1, ligandpos, ligandnom})]
] (* for successful interactions make the receptor unavailable for further binding; associate the unavailability index
of 1, ligand-type and ligand index *)
interAct[specie_, specieind_, membraneind_, interactingspecies : {{_, _} ..},
ligandname_] :=
Module[{interactionProb, successpos, successInt, membraneIndUpdate,
successligand, specielist = specie, index = specieind},
interactionProb = Table[RandomReal[], Length@interactingspecies]; (* lets generate some probabilites equal to how many
interactions are observed *)
successpos = Position[interactionProb, _?(# > 0.5 &)]; (* finding positions where the probability is greater than a
defined value *)
successInt = interactingspecies[[Flatten@successpos]]; (* successful ligand-receptor interaction pairs *)
membraneIndUpdate =
Fold[bindingEvent[#1, #2, ligandname] &, {membraneind, ## & @@
successInt}]; (* run bindingEvent module for successful interactions *)
successligand = # & @@@ successInt;(* all the ligands that have successfully interacted *)
specielist =
Fold[Function[{list, index},
list /. {y_, _, {_, _}} /; y == index :> Sequence[]], {specielist, ## & @@ successligand}]; (* deleting successful
ligands from the ligandlist *)
KeyDropFrom[index, successligand]; (* deleting ligands from the ligand index *)
{specielist, index, membraneIndUpdate} (* sending the list of updated ligandlist, ligandindex and membraneindex *)
] /; Count[membraneind, 0, 3] > 0 && Length@specieind > 0 (* execute the module only if a free receptor is present *)
for unbinding of the ligands from the receptors
unbindingMechanics[membraneind_, ligandlist_, ligandind_,ligandname_?StringQ] :=
Module[{membraneInd = membraneind, ligandList = ligandlist, ligandInd = ligandind, boundSpecies, probability,
position, dissocligand},
boundSpecies =
Cases[membraneInd,
PatternSequence[_Integer -> {{p1_, p2_}, 1, y_,
ligandname}] :> {y, {p1, p2}}]; (* finding all the bound ligands from membraneindex and placing their index together
with receptor position *)
probability = Table[RandomReal[], Length@boundSpecies]; (* lets generate probabilities equal to number of bound species *)
position = Position[probability, _?(# > 0.95 &)]; (* find positions of successful dissociations *)
dissocligand = boundSpecies[[All, 1]][[Flatten@position]]; (* find all the ligands that have been dissociated *)
membraneInd =
Replace[membraneInd,
PatternSequence[
x_Integer -> {{p1_, p2_}, 1, Alternatives @@ dissocligand,
ligandname}] :> (x -> {{p1, p2}, 0, None, "type"}), 1]; (* freeing up the membrane receptors available for successful
dissociations *)
ligandInd = KeySort@Append[ligandInd, Thread[dissocligand -> 1]]; (* add the dissociated ligands back to the ligand index
and then put them in ascending order *)
ligandList =
SortBy[Fold[Insert[#1, {#2[[1]], 0, #2[[2]]}, -1] &, ligandList,
boundSpecies[[Flatten@position]]] , First]; (* adding the ligand back to the ligand list together with its position *)
{membraneInd, ligandList, ligandInd} (* sending the updated {membraneindex, ligandlist,ligandindex} out of module *)
] /; Count[membraneind, ligandname, 3] > 0 (* run only if the particular ligand bound *)
for the production of new ligands
productionEvent[diff_?(# > 0 &), list_, ind_, count_] :=
Module[{particlelist = list, particleind = ind, newpoints, newindices, countupdate},
newpoints = Table[RandomPoint[\[ScriptCapitalR]], diff]; (* generate new points *)
newindices = Range[#, # + diff][[2 ;;]] &@count; (* generating the indices of the newly added points *)
countupdate = count + diff; (* increase the counter so that new particles produced do not interfere with existing ones *)
MapThread[AppendTo[particlelist, {#1, 0, #2}] &, {newindices, newpoints}]; (* add the new points to the existing ones *)
AppendTo[particleind, Thread[newindices -> 0]]; (* add the new particle indices to the index list *)
{particlelist, particleind, countupdate}
]
produceParticles[nodallis_, leftylis_, nodalin_, leftyin_, membraneind_, countnod_, countleft_, listprod_] :=
Module[{nodalthread = nodallis, leftythread = leftylis, nodalBindex = nodalin, leftyBindex = leftyin, boundnodal,
boundlefty, diff, countnodal = countnod, countlefty = countleft, listp = listprod},
boundnodal = Count[membraneind, "nodal", 3];(* count the number of bound nodal particles *)
AppendTo[listp, boundnodal]; (* add to the list that accounts for the production delay *)
If[Length@listp == 10,
(diff = (#2 - #1) & @@ Take[listp, 2];
boundlefty = Count[membraneind, "lefty", 3];
{{nodalthread, nodalBindex, countnodal}, {leftythread, leftyBindex, countlefty}} =
MapThread[(productionEvent[##]) /. productionEvent[x_, y__] :> {y} &, {{diff, diff}, {nodalthread, leftythread},
{nodalBindex, leftyBindex}, {countnodal, countlefty}}];
listp = Delete[listp, 1];)]; (* when the size of the list is equal to some integer number execute the code, which in essence
means a delay before the first production event *)
{nodalthread, nodalBindex, leftythread, leftyBindex, countnodal, countlefty, listp}
]
for degradation of ligands
degradationParticles[particleind_, particlelist_] := Module[{randProb, successpos, ind = particleind, list = particlelist},
randProb = Table[RandomReal[], Length@particlelist]; (* we generate random probabilities equal to number of free ligands *)
successpos = Flatten@Position[randProb, _?(# > 0.99999 &)]; (* find positions of successful kills *)
successpos = successpos /. patt : {__Integer} :> Partition[patt, 1];
{ind, list} = Delete[#, successpos] & /@ {ind, list}; (* delete ligands from the list and index *)
{ind, list}
] /; Length@particleind > 0 (* if free ligands exist then execute module *)
The section below is what ties all the helper functions together i.e. to make sure the sequential execution
BrownianSimulation[nodalp_, nodalBoundaryIndex_, leftyp_, leftyBoundaryIndex_, membraneind_, membranep_, countn_, countl_,
listnp_] :=
Module[{nodalcopy, nodal, lefty, leftycopy, nodalthreadlist, leftythreadlist, interactingNodalReceptor,
interactingLeftyReceptor, leftyind, nodalind, membraneIndex, membranepoints, boundnodal, boundlefty,
countnod = countn, countleft = countl, listprod = listnp},
nodalind = nodalBoundaryIndex; (* initial boundary configuration of nodal in the system *)
leftyind = leftyBoundaryIndex; (* initial configuration of lefty in the system *)
nodal = nodalp; (* save initial configuration of nodal*)
lefty = leftyp;
nodalcopy = nodal;
leftycopy = lefty;
(* ------ brownian step ------ *)
{nodal, lefty} = Map[If[Length@# != 0, BrownianFunc[#], #] &, {nodal, lefty}];
(* brownian step for receptors *)
{membranepoints, membraneIndex} = BrownianWalkMembrane[membranep, membraneind];
(* ------ ligand spatial checks ------ *)
{nodalthreadlist, leftythreadlist} = MapThread[ligandCheck, {{nodal, lefty}, {nodalcopy, leftycopy}, {nodalind,leftyind}}];
nodalind = Association@Map[#1[[1]] -> #[[2]] &, nodalthreadlist];
leftyind = Association@Map[#1[[1]] -> #[[2]] &, leftythreadlist];
(* ------ check for possible ligand-receptor interactions ------ *)
{interactingNodalReceptor, interactingLeftyReceptor} = Map[(nearestFunction[#, membraneIndex, membranepoints] /.
nearestFunction[__] :> {}) &, {nodalthreadlist,leftythreadlist}];
(* biasing a common receptor interacting with nodal and lefty at the same time *)
{interactingNodalReceptor, interactingLeftyReceptor} =
(biasSharedReceptors[interactingNodalReceptor, interactingLeftyReceptor]) /. biasSharedReceptors[pattern__] :> {pattern};
(* ------ binding mechanics ------ *)
{nodalthreadlist, nodalind, membraneIndex} = (interAct[##] & @@ {nodalthreadlist, nodalind, membraneIndex,
interactingNodalReceptor, "nodal"}) /. interAct[patt__, y_, z_] :> {patt};
{leftythreadlist, leftyind, membraneIndex} = (interAct[##] & @@ {leftythreadlist, leftyind, membraneIndex,
interactingLeftyReceptor, "lefty"}) /. interAct[patt__, y_, z_] :> {patt};
(* ------ degradation ------ *)
{nodalind, nodalthreadlist} = (degradationParticles[nodalind, nodalthreadlist]) /. degradationParticles[x__] :> {x};
{leftyind, leftythreadlist} = (degradationParticles[leftyind, leftythreadlist]) /. degradationParticles[x__] :> {x};
(* ------ production of particles ------ *)
{nodalthreadlist, nodalind, leftythreadlist, leftyind, countnod, countleft, listprod} =
produceParticles[nodalthreadlist, leftythreadlist, nodalind, leftyind, membraneIndex, countnod, countleft, listprod];
(* ------ unbinding mechanics ------ *)
{membraneIndex, nodalthreadlist, nodalind} = (unbindingMechanics[membraneIndex, nodalthreadlist,
nodalind, "nodal"]) /. unbindingMechanics[patt__, y_] :> {patt};
{membraneIndex, leftythreadlist, leftyind} = (unbindingMechanics[membraneIndex, leftythreadlist,
leftyind, "lefty"]) /. unbindingMechanics[patt__, y_] :> {patt};
(* export the results out *)
{nodalthreadlist, nodalind, leftythreadlist, leftyind, membraneIndex, membranepoints, countnod, countleft, listprod}
]
the function below draws the spatiotemporal evolution
graphState[membraneind_, nodalthread_, leftythread_, boundnod_,boundlef_] :=
Graphics[{{Gray, Dashed, Line[{{-1, -1}, {1, -1}, {1, 1}, {-1, 1}, {-1, -1}}]}, {Black, Opacity[0.2], Circle[{0, 0}, 0.75]},
{PointSize[Medium], Opacity[0.6], Darker@Purple, Point@membraneind[[All, 2, 1]]}, {Darker@Blue, PointSize[Medium], Opacity[0.6],
Point@nodalthread[[All, 3]]}, {Red, Opacity[0.6], PointSize[Medium], Point@leftythread[[All, 3]]}, {Darker@Green,
PointSize[Large], Opacity[0.4], Point@boundnod}, {Darker@Red, PointSize[Large], Opacity[0.4], Point@boundlef}},
ImageSize -> Large]
(* draws the system state *)
I use the code below to first generate initial configuration of the system and then run BrownianSimulation multiple times using Nest and drawing the system state using graphState
sim = With[{bounds = 0.25, numnodalparticles = 40, numleftyparticles = 30, center = {0, 0}, cellradius = 0.75,
numreceptors = 100, nSteps = 3000},
Module[{nodallist, nodalBoundaryindex, nodalpts, leftylist, leftyBoundaryindex, leftypts, membranepts, membraneindex,
countnod, countleft, listprod = {}, temp, g, boundn, boundp},
(* we generate the initial configuration here *)
{nodallist, nodalBoundaryindex, nodalpts} = initialParticleConfig["nodal", {-bounds, bounds}, numnodalparticles];
countnod = Length@nodalBoundaryindex;
{leftylist, leftyBoundaryindex, leftypts} = initialParticleConfig["lefty", {-bounds, bounds}, numleftyparticles];
countleft = Length@leftyBoundaryindex;
{membranepts, membraneindex} = membraneReceptorConfig[center, cellradius, numreceptors];
(* printing the intial configuration *)
Print@Graphics[{{Gray, Dashed, Line[{{-1, -1}, {1, -1}, {1, 1}, {-1, 1}, {-1, -1}}]}, {Black, Opacity[0.2],
Circle[center, cellradius]}, {PointSize[Medium], Opacity[0.6], Darker@Purple, Point@membranepts}, {Darker@Blue,
PointSize[Medium], Opacity[0.6], Point@nodalpts}, {Red, Opacity[0.6], PointSize[Medium], Point@leftypts}},
ImageSize -> Large];
Monitor[Reap[Nest[(temp = BrownianSimulation @@ #;
boundn = Cases[temp[[5]], patt : PatternSequence[_ -> {{_, _}, _, _, "nodal"}] :> patt[[2, 1]], 2];
boundp = Cases[temp[[5]], patt : PatternSequence[_ -> {{_, _}, _, _, "lefty"}] :> patt[[2, 1]], 2];
Sow[{Length@boundn + Length@temp[[1]], Length@boundp + Length@temp[[3]]}];
g = graphState[temp[[5]], temp[[1]], temp[[3]], boundn, boundp];
temp[[1]] = Association@Map[#[[1]] -> {"nodal", #[[3]]} &, temp[[1]]];
temp[[3]] = Association@Map[#[[1]] -> {"lefty", #[[3]]} &, temp[[3]]];
temp) &, {nodallist, nodalBoundaryindex, leftylist,
leftyBoundaryindex, membraneindex, membranepts, countnod,
countleft, listprod}, nSteps]],
g] // Quiet
]
];
Please note that the current system has a lot of free parameters that need to be fixed from experiments. Only then we will know whether the system yields any stable spatio-temporal turing pattern
 Attachments:
Attachments:


