This reply is about importing structures from CIF files.
I'll be using this CIF file of Rutile as an example and of course the CifImport package for import. But you will NEED to add something to the end of the CIF file. The last line is currently this: O 0.30530 0.30530 0.00000 You can just type "end" into the following line, or anything else you like; even an additional blank line will help. I'm sorry about this, it's a glitch in the way Import reads the file, so from my end, it could only be fixed by reading the file as a plain table instead.
CIF files are widely used for crystal structures, so if you're looking for a structure, you can usually find a CIF in a database somewhere. But you will see that the crystal structure in a CIF file is impossible to read for most humans, and not entirely trivial to import either. Exporting to CIF also requires extra work that is outside the scope of my packages. On the other hand, CIF files contain lots of metadata such as symmetry properties of the structure, information about the samples themselves, and citation information.
CIF is essentially a markup format where you have a name for each dataset and then the actual data. Mathematica can import our example CIF file and will return a list of rules linking the names and data of each dataset:
Import["Rutile.cif"]
So why do I keep saying that CIF files are not so trivial? Well, let's look at the actual file. You can open it in a text editor or via FilePrint.
Roughly the first half of the file is nicely readable for a human, you get citation information with journal and author names and years and pages and whatnot. Sometimes you'll get information on where the sample was found, or how it was synthesized. Then you get the lattice vectors in terms of their lengths and angles, and the space group. All of that is fine and great to have. But the actual crystal structure data is not so easy, look at this:
loop_
_space _group _symop _operation _xyz
'x,y,z'
'-y,-x,z'
'y,x,-z'
'1/2+y,1/2-x,1/2-z'
'1/2-y,1/2+x,1/2+z'
'1/2+x,1/2-y,1/2+z'
'1/2-x,1/2+y,1/2-z'
'x,y,-z'
'-x,-y,z'
'y,x,z'
'-y,-x,-z'
'1/2-y,1/2+x,1/2-z'
'1/2+y,1/2-x,1/2+z'
'1/2-x,1/2+y,1/2+z'
'1/2+x,1/2-y,1/2-z'
'-x,-y,-z'
loop_
_atom _site _label
_atom _site _fract _x
_atom _site _fract _y
_atom _site _fract _z
Ti 0.00000 0.00000 0.00000
O 0.30530 0.30530 0.00000
Spoiler alert: This structure, rutile, has 2 Ti and 4 O atoms, and it's a popular crystallization choice among metal dioxides. But the "atom_site" datasets only give us one atom of each type! What's going on here? Is it alchemy? Well, not quite. In addition to these 2 atoms, we also get all symmetry operations of the cell. If we assume for a moment that we have the complete cell with all 6 atoms, we could apply any of these symmetry transformations and we'd end up with the same structure. Atoms would be mapped onto other atoms of the same type. Back to our 2 atoms in the file: If we apply all symmetry operations to these 2 atoms, we will sometimes map them onto themselves, but at other times they will wind up on the positions of their missing friends. And this is how we can construct the complete crystal structure from a CIF file.
So let's import the structure "properly", with CifImport:
Needs["CifImport`"];
CifImport["Rutile.cif"]
... which gives us the following output:
{
<|"lattice"->{{4.59373`,0,0},{0,4.59373`,0},{0,0,2.95812`}},"atomcoords"->{{0.`,0.`,0.`},{0.3053`,0.3053`,0.`}},"atomtypes"->{1,2},"chemical"->{"Ti","O"},"name"->"Rutile","file"->"E:\\Rutile.cif","comment"->"atoms of the asymmetric unit with NONchemical types"|>,
<|"lattice"->{{4.59373`,0,0},{0,4.59373`,0},{0,0,2.95812`}},"atomcoords"->{{0.`,0.`,0.`},{0.5`,0.5`,0.5`},{0.30529999999999996`,0.30529999999999996`,0.`},{0.6947000000000001`,0.6947000000000001`,0.`},{0.8053`,0.19469999999999998`,0.5`},{0.19469999999999998`,0.8053`,0.5`}},"atomtypes"->{1,1,2,2,2,2},"chemical"->{"Ti","O"},"name"->"Rutile","file"->"E:\\Rutile.cif","comment"->"complete cell with chemical types"|>,
<|"lattice"->{{4.59373`,0,0},{0,4.59373`,0},{0,0,2.95812`}},"atomcoords"->{{0.`,0.`,0.`},{0.3053`,0.3053`,0.`}},"atomtypes"->{1,2},"chemical"->{"Ti","O"},"name"->"Rutile","file"->"E:\\Rutile.cif","comment"->"atoms of the asymmetric unit with chemical types"|>,
<|"lattice"->{{4.59373`,0,0},{0,4.59373`,0},{0,0,2.95812`}},"atomcoords"->{{0.`,0.`,0.`},{0.5`,0.5`,0.5`},{0.30529999999999996`,0.30529999999999996`,0.`},{0.6947000000000001`,0.6947000000000001`,0.`},{0.8053`,0.19469999999999998`,0.5`},{0.19469999999999998`,0.8053`,0.5`}},"atomtypes"->{1,1,2,2,2,2},"chemical"->{"Ti","O"},"name"->"Rutile","file"->"E:\\Rutile.cif","comment"->"complete cell with chemical types"|>
}
We get a list of Associations, where each Association represents one crystal structure. Why are there multiple structures? Well, read the "comment" entries: CifImport will give you the complete structure, but also the structure recorded in the file (which is sometimes called an asymmetric unit cell). Also, some structures need multiple atoms of the same type even within the asymmetric unit, and CIF files usually tag these atoms with labels that are suddenly no longer just the chemical species - so that's where the "nonchemical types" come in.
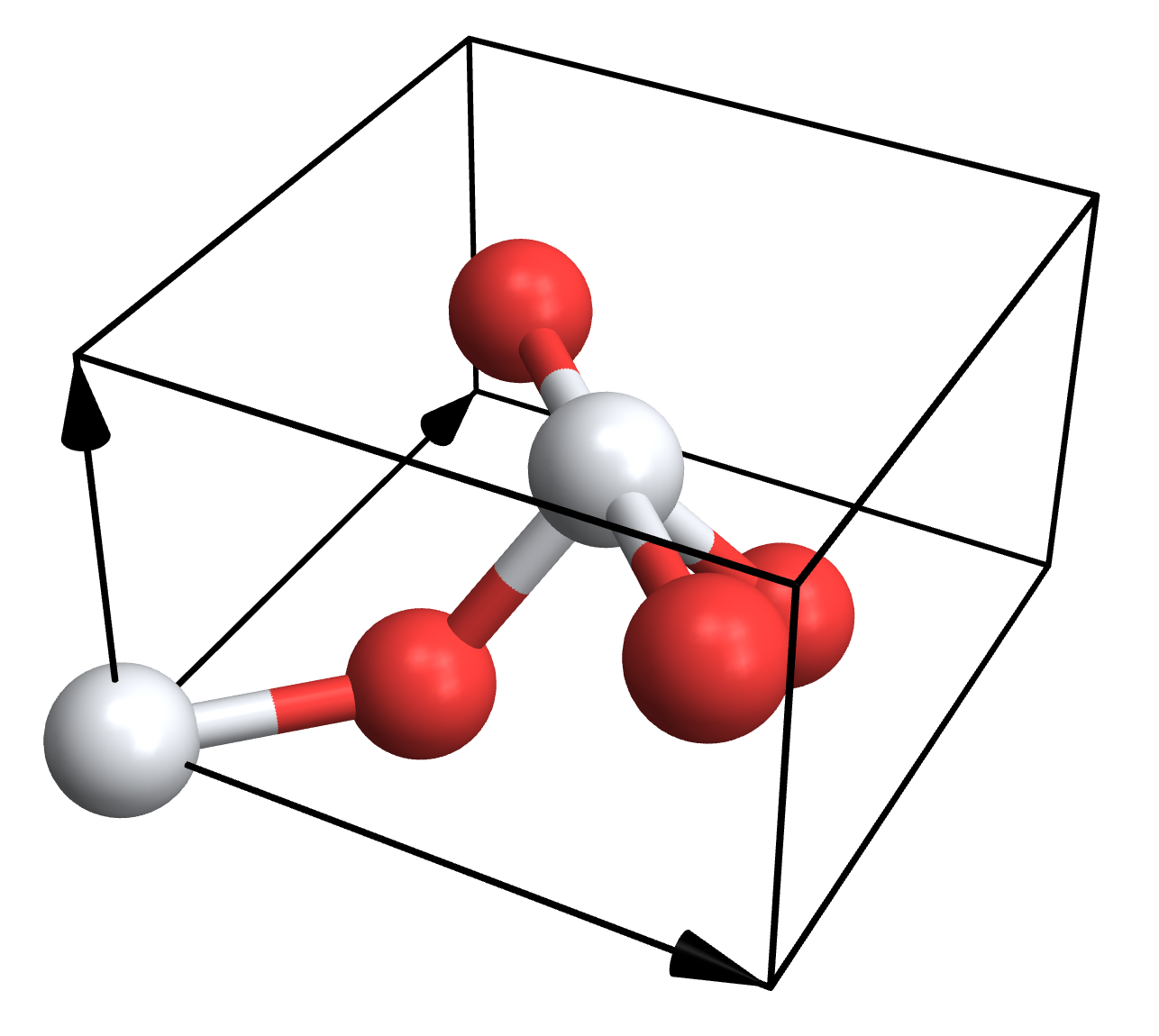
The structure you'll usually be interested in is the last one, which has all the atoms and chemical types. Let's just hook up the import with Crystallica really quickly:
Needs["Crystallica`"];
Needs["CifImport`"];
cifPlot[file_,options___]:=With[{data=Last[CifImport[file]]},
CrystalPlot[data["lattice"],data["atomcoords"],data["atomtypes"],options,AtomCol->data["chemical"]]];
cifPlot["Rutile.cif"]
And we get a plot!
If you wanted to export a structure to CIF format, you would definitely need code that finds the symmetries of your structure, and that can also return the actual name of the space group. You can write that, but you will find that various programs already exist that can export to CIF format, so you might be better off using one of those instead.


