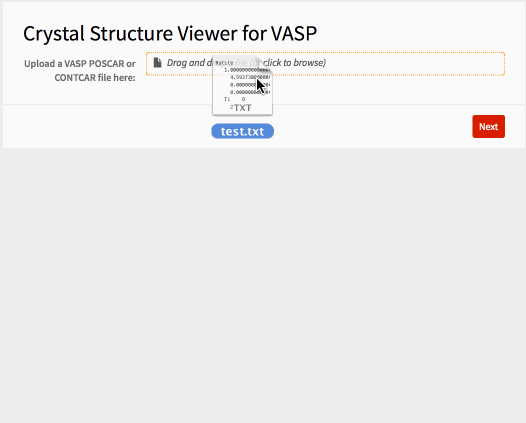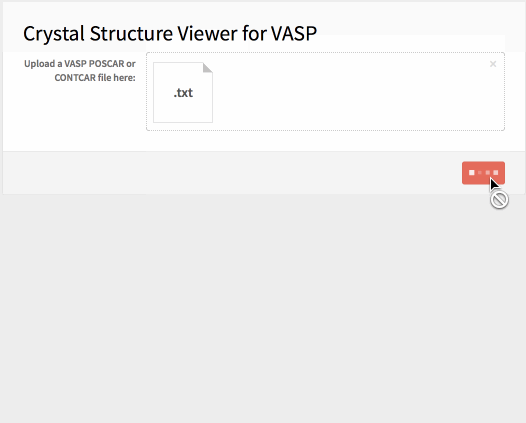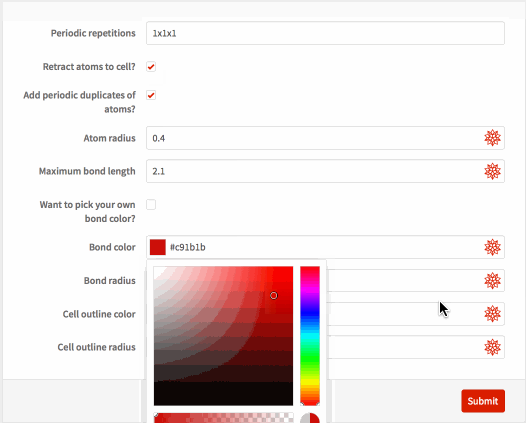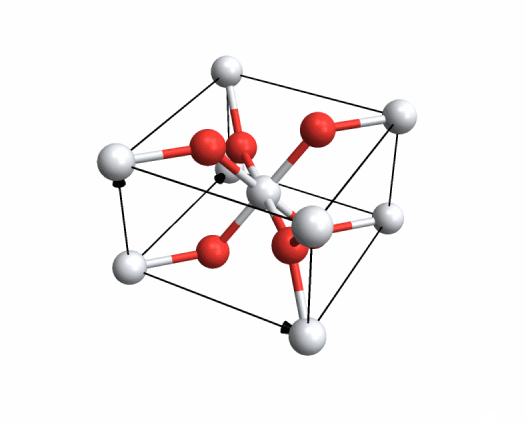
You may remember that I've already posted packages for crystal structure plots. I just wanted to dump a piece of code here that's based on these packages and will let you plot crystal structures from VASP input/output files on a webpage... in case anyone else is using VASP.
For anyone who wants to try the app but doesn't have a VASP file handy, here's a sample you can copy and paste into a plain text file:
Rutile
1.00000000000000
4.5937300000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 4.5937300000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 2.9581200000000000
Ti O
2 4
Direct
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.5000000000000000 0.5000000000000000 0.5000000000000000
0.3053000000000000 0.3053000000000000 0.0000000000000000
0.6947000000000000 0.6947000000000000 0.0000000000000000
0.8053000000000000 0.1947000000000000 0.5000000000000000
0.1947000000000000 0.8053000000000000 0.5000000000000000
The actual code is below the image. Have fun!
CloudDeploy[FormFunction[{
{{"file", "Upload a VASP POSCAR or CONTCAR file here:"} -> <|
"Interpreter" -> "UploadedFile"|>}, {
{"sysdim", "Periodic repetitions"} -> <|
"Interpreter" ->
Restricted[
DelimitedSequence["Integer",
" " | "," | ";" | "/" | "x" | "X"], 3], "Input" -> "1x1x1"|>,
{"retractq", "Retract atoms to cell?"} -> <|
"Interpreter" -> "Boolean", "Input" -> False|>,
{"addq", "Add periodic duplicates of atoms?"} -> <|
"Interpreter" -> "Boolean", "Input" -> False|>,
{"atomrad", "Atom radius"} -> <|"Interpreter" -> "ComputedReal",
"Input" -> 0.4|>,
{"bonddist", "Maximum bond length"} -> <|
"Interpreter" -> "ComputedReal", "Input" -> 2.1|>,
{"monobond", "Want to pick your own bond color?"} -> <|
"Interpreter" -> "Boolean", "Input" -> False|>,
{"bondcol", "Bond color"} -> <|"Interpreter" -> "ComputedColor",
"Input" -> GrayLevel[.8]|>,
{"bondrad", "Bond radius"} -> <|"Interpreter" -> "ComputedReal",
"Input" -> 0.1|>,
{"linecol", "Cell outline color"} -> <|
"Interpreter" -> "ComputedColor", "Input" -> Black|>,
{"linerad", "Cell outline radius"} -> <|
"Interpreter" -> "ComputedReal", "Input" -> 0.02|>
}},
Module[{file, sysdim, retractq, addq, atomrad, bonddist, monobond,
bondcol, bondrad, linecol, linerad,
data, speciesQ, atomcol, chemoffset, chgoffset, lattscale,
lattvec, coord, conf, coordP, confP, new, tubearrow, lines,
atoms, tuples, bonds},
file = #file;
sysdim = #sysdim;
retractq = #retractq;
addq = #addq;
atomrad = #atomrad;
bonddist = #bonddist;
monobond = #monobond;
bondcol = #bondcol;
bondrad = #bondrad;
linecol = #linecol;
linerad = #linerad;
(*import: *)
data = Import[file, "Table"];
(*offset if chemical species are given: *)
speciesQ = MatchQ[data[[6]], {_String ..}];
atomcol[type_] :=
If[speciesQ, ColorData["Atoms", data[[6, type]]],
ColorData[97, type]];
chemoffset = If[speciesQ, 1, 0];
(*offset for missing "Selective Dynamics" line as is common in \
CHG/CHGCAR: *)
chgoffset =
If[TrueQ[Length[data[[chemoffset + 8]]] > 0] &&
MatchQ[data[[chemoffset + 8, 1]], _?NumericQ], -1, 0];
(*lattice vectors and atoms: *)
lattvec = data[[3 ;; 5]];
lattscale = data[[2]];
If[TrueQ[Length[lattscale] == 1], lattscale = lattscale[[1]]];
If[TrueQ[Sign[lattscale] == -1],
lattscale = (-lattscale/Det[lattvec])^(1/3)];
lattvec =
If[TrueQ[Length[lattscale] == 3],
N[lattscale[[#]]*lattvec[[#]]] & /@ Range[3],
N[lattscale*lattvec]];
conf =
Flatten[Join[
ConstantArray[#, data[[chemoffset + 6, #]]] & /@
Range[Length[data[[chemoffset + 6]]]]]];
coord =
N[data[[chemoffset + chgoffset +
9 ;; (chemoffset + chgoffset + 8 +
Total[data[[chemoffset + 6]]]), 1 ;; 3]]];
(*reprojection for cartesian coordinates: *)
If[TrueQ[Length[data[[chemoffset + chgoffset + 8]]] > 0] &&
StringMatchQ[ToString[data[[chemoffset + chgoffset + 8, 1]]],
"c*" | "k*", IgnoreCase -> True],
If[TrueQ[Length[lattscale] == 3], coord = #*lattscale & /@ coord,
coord = coord*lattscale];
coord = coord.Inverse[lattvec]];
{coordP, confP} = {coord, conf};
(*retraction, moving all atoms back into the cell: *)
If[retractq, With[{retracttol = 10^-4},
coordP =
Partition[
If[# > (1 - retracttol), # - 1, #] & /@
Flatten[# - Floor[#] & /@ coordP], 3];
new = DeleteDuplicates[Transpose[{coordP, confP}],
(Norm[
Round[#1[[1]], retracttol] - Round[#2[[1]], retracttol] -
Floor[Round[#1[[1]], retracttol]] +
Floor[Round[#2[[1]], retracttol]]] <
retracttol) && (#1[[2]] == #2[[2]]) &];
{coordP, confP} = Transpose[new]]];
(*periodic repetition: *)
coordP =
Flatten[Table[(# + {a, b, c}) & /@ coordP, {a, 0,
sysdim[[1]] - 1}, {b, 0, sysdim[[2]] - 1}, {c, 0,
sysdim[[3]] - 1}], 3];
confP = Flatten[ConstantArray[confP, Times @@ sysdim]];
(*add peridic duplicates if desired: *)
If[TrueQ[addq],
new = Transpose[{coordP, confP}];
new =
Join[new, # + {{sysdim[[1]], 0, 0}, 0} & /@
Select[new,
Abs[#[[1, 1]]] < 0.01 &], # + {{-sysdim[[1]], 0, 0}, 0} & /@
Select[new, Abs[#[[1, 1]]] > 0.99*sysdim[[1]] &]];
new =
Join[new, # + {{0, sysdim[[2]], 0}, 0} & /@
Select[new,
Abs[#[[1, 2]]] < 0.01 &], # + {{0, -sysdim[[2]], 0}, 0} & /@
Select[new, Abs[#[[1, 2]]] > 0.99*sysdim[[2]] &]];
new =
Join[new, # + {{0, 0, sysdim[[3]]}, 0} & /@
Select[new,
Abs[#[[1, 3]]] < 0.01 &], # + {{0, 0, -sysdim[[3]]}, 0} & /@
Select[new, Abs[#[[1, 3]]] > 0.99*sysdim[[3]] &]];
{coordP, confP} = Transpose[new];
];
(*cell lines: *)
tubearrow[{tail_, head_}] :=
With[{scale = .5*Sqrt[Mean[Norm /@ lattvec]*linerad]},
Tube[{tail, head - 4*scale*Normalize[head - tail],
head - 4*scale*Normalize[head - tail], head}, {linerad,
linerad, scale, 0}]];
lines = {linecol, Tube[#.lattvec, linerad] & /@ {
{{0, 0, #}, {1, 0, #}, {1, 1, #}, {0, 1, #}, {0,
0, #}} & /@ {0, 1},
{{0, 0, #} & /@ {0, 1}, {1, 0, #} & /@ {0,
1}, {1, 1, #} & /@ {0, 1}, {0, 1, #} & /@ {0, 1}}
}, tubearrow[{{0, 0, 0}, #}] & /@ lattvec};
(*atoms: *)
atoms =
Tooltip[{atomcol[confP[[#]]],
Sphere[coordP[[#]].lattvec, atomrad]}, #] & /@
Range[Length[confP]];
(*bonds: *)
tuples =
Select[Subsets[Range[Length[confP]], {2}],
Norm[(coordP[[#]].lattvec)[[1]] - (coordP[[#]].lattvec)[[2]]] <
bonddist &];
bonds = If[TrueQ[tuples == {}], {},
If[monobond,
{bondcol, Tube[coordP[[#]].lattvec, bondrad]},
Table[{atomcol[confP[[#[[ii]]]]],
Tube[{coordP[[#[[ii]]]].lattvec,
Total[coordP[[#]].lattvec]*.5}, bondrad]}, {ii, 1, 2}]
] & /@ tuples];
(*return the plot: *)
Graphics3D[{lines, bonds, atoms}, ImageSize -> Large,
BaseStyle -> {Specularity[Gray, 100]}, Boxed -> False,
SphericalRegion -> True, Lighting -> "Neutral"]
]
&, "PNG",
AppearanceRules -> <|
"Title" -> "Crystal Structure Viewer for VASP"|>],
Permissions -> "Public"]


